


Sommaire
- Le 6 Sigma et l'Excellence Opérationnelle. Juste du bon sens ?
- Combien de valeurs sont nécessaires pour avoir un échantillon représentatif ?
- Exploiter la donnée pour optimiser le pilotage d'un procédé
- Statistical modeling: The need for a reliable approach to improve process knowledge and understanding
- Bayesian approach in cosmetical research : Application to a meta-analysis on the anti-pigmenting effect of vitamin C
- Comparabilité, équivalence, similarité... Comment les statistiques peuvent nous aider à en faire la démonstration. Et bientôt la fin d'un "blind test" pour les autorités de santé et les industriels
- Le maintien du statut validé, une étape du cycle de validation
- Stratégie de validation des procédés et mise en application de l'Annexe 15 des BPF et des guidances FDA. Vérification continue des procédés (CPV)
Depuis quelques années déjà, l’industrie pharmaceutique, qui jusque-là très frileuse à cause d’une pression règlementaire stricte et peu adaptée à la gestion des changements, doit régulièrement optimiser et modifier ses procédés de fabrications, rénover ses installations de production ou pour améliorer la flexibilité et l’agilité, réaliser des transferts de procédés sur de nouveaux sites de production.

En effet, poussés par les innovations technologiques et par la nécessité d’améliorer la performance et la maitrise des procédés, les industriels s’engagent dans des plans d’amélioration qui vont de facto impacter le statut validé des procédés et les dossiers d’autorisation de mise sur le marché, s’accompagnant parfois d’études cliniques complémentaires et donc de procédures règlementaires longues et complexes.
Les autorités de santé ont également pris conscience de l’intérêt de cette évolution participant de manière très significative à la sécurité et à la disponibilité des médicaments.
C’est pourquoi, la règlementation pharmaceutique évolue régulièrement avec l’émission de guides ou la mise à jour des GMPs intégrant, entre autre, le concept de “cycle de vie du produit”, l’évaluation et la gestion des risques, le “Quality by design”, la vérification en continu des procédés.
On retrouve entre autre :
- ICH Q5E: Note for guidance on biotechnological/biological products subjected to changes in their manufacturing process (CPMP/ICH/5721/03)
- ICH Q8 : Pharmaceutical Development (EMA/CHMP/ICH/167068/2004)
- ICH Q9 : Quality Risk Management (EMA/CHMP/ICH/24235/2006)
- ICH Q10 : Pharmaceutical quality system (EMA/CHMP/ICH/214732/2007)
- ICH Q11 : Development and manufacture of drug substances (chemical entities and biotechnological/biological entities, EMA/CHMP/ICH/425213/2011.
Plus récemment la guideline ICH Q12 : Technical and regulatory considerations for pharmaceutical product lifecycle management, qui était en consultation publique jusqu’en décembre 2018 et devant être adoptée fin 2019 concrétise le souhait des autorités de santé de simplifier les procédures de règlementaires en vue de favoriser l’innovation et l’amélioration continue.
Toute modification de procédé intervenant sur un procédé commercialisé doit être soutenue par une étude de validation permettant de démontrer la maitrise et la reproductibilité du procédé modifié mais également complétée par une étude de comparabilité afin de démontrer que la qualité du produit obtenue après modification du procédé est comparable à celle du produit avant modification. Cette comparabilité permettra ainsi de limiter voire d’éviter les études cliniques pour un changement post approbation.
Un des éléments prépondérants pour démontrer cette comparabilité est l’utilisation des statistiques. Encore faut-il savoir quels outils statistiques utiliser et concevoir les études de manière appropriée. Taille de l’échantillonnage, nombre de répétitions, niveaux de risques… sont autant de paramètres difficiles à définir pour des non statisticiens. De plus en plus d’entreprises possèdent aujourd’hui leur propre département “statistiques” ou font appel à des sociétés d’expertise en statistiques. Mais là encore, on assiste souvent à des difficultés liées à des incompréhensions entre deux expertises différentes : les experts procédé/produits et les statisticiens.
Il n’est pas facile pour le demandeur de l’étude d’exprimer clairement ses besoins. Face à une expression du besoin floue, le statisticien ne peut pas fournir une réponse adaptée sans comprendre de façon approfondie la problématique. On assistera alors à une analyse statistique conduisant au mieux à des résultats ininterprétables ou au pire à une interprétation erronée comme par exemple, la non comparabilité des résultats alors que les résultats sont comparables ou l’inverse.
C’est pourquoi, un réel travail collaboratif entre les 2 entités et une compréhension mutuelle des besoins et contraintes sont primordiaux. On observe également au niveau de l’inspectorat ou des évaluateurs des divergences d’approches, des incompréhensions et un langage ou vocabulaire hétérogène.
On peut imaginer aisément les situations difficiles en inspection pour présenter de telles études si chaque partie ne possède pas un langage commun et une expertise du sujet. Bien évidemment les impacts seront inversement proportionnels au niveau d’incompréhension ou d’expertise entre les différents acteurs.
C’est pourquoi, en mars 2017, l’EMA a publié un draft nommé : “Reflection paper on statistical methodology for the comparative assessment of quality attributes in drug development“. Ce draft a été en consultation publique pendant 1 an pour permettre aux industries d’interagir avec l’EMA au travers du comité scientifique SAWP (Scientific Advice Working Party) sur le contenu mais aussi pour pouvoir proposer des méthodes alternatives non présentées dans ce document. A l’issue de cette période de consultation, l’EMA a organisé, en mai 2018, un workshop avec les industriels afin de partager sur le contenu et ses implications dans l’industrie.
En 2017, A3P a créé un GIC “Statistiques” afin, dans un premier temps, de commenter ce draft puis ensuite de préparer un guide. Début 2018, les membres du GIC ont soumis leurs commentaires puis en mai 2018, un représentant du GIC a participé au nom d’A3P au workshop organisé par l’EMA.
Cet article a pour objectif de faire une synthèse des points importants du draft (en cours de discussion à l’EMA avec une sortie définitive prévue fin 2019), des commentaires soumis par A3P et terminera par une conclusion incluant la suite des travaux du GIC statistiques.
1. Résumé du draft “Reflection paper on statistical methodology for the comparative assessment of quality attributes in drug development”
La comparaison des données des attributs qualité du produit est incontournable que ce soit pendant les phases de développement d’un nouveau produit ou tout au long du cycle de vie d’un médicament commercialisé. Le souhait des autorités de santé, au travers de ce document, est d’établir un langage commun et de renforcer la compréhension au sein des différents experts intervenant dans les études de comparaison des attributs qualité.
Ce document a donc pour objectif de présenter des approches statistiques permettant de comparer des données liées à la qualité du produit ou en d’autres termes les caractéristiques produit dans les 3 cas suivants :
- changements de procédé et/ou de méthode analytique portant sur un produit commercialisé,
- comparaison d’un candidat biosimilaire à un médicament de référence
- comparaison d’un générique à un médicament de référence.
Dans le cadre de cet article, la synthèse porte principalement sur le premier cas : changement de procédé portant sur un produit commercialisé. En effet, les membres du GIC n’ayant pas suffisamment d’expertise liée aux biosimilaires et génériques nous ne nous sommes pas permis d’analyser et commenter ces parties.
L’objectif d’une étude de comparaison est de démontrer que deux produits sont comparables ou que deux profils de contrôle peuvent être considérés comme similaires.
Une des difficultés rencontrées lorsqu’on veut comparer deux populations est la définition des critères d’acceptation puisque la taille de l’échantillon à évaluer est faible. En effet, que ce soit pendant les phases de développement ou dans le cadre d’un changement de procédé, le nombre de lots servant à l’étude de comparabilité (lots de validation ou lots de développement) est limité. Pour avoir un échantillon représentatif, il faudrait produire à minima une dizaine de lots tout en intégrant la variabilité intrinsèque des procédés (effets campagnes, variabilité des matières premières…). Pour les industriels, le coût et le temps nécessaire à une telle étude est impossible à assumer puisque ce sont des lots à échelle industrielle et la plupart du temps non commercialisables. Les autorités de santé en ont conscience et ne requièrent donc pas un nombre de lot minimum car ce serait un frein à l’innovation, l’amélioration continue renforçant la sécurité des produits et à la mise à disposition des médicaments. C’est pourquoi, ce document insiste beaucoup sur les approches statistiques basées sur les petits échantillons.
1.1. Comparaison de données dans le cadre d’un changement de procédé commercialisé
La comparaison des attributs qualité du produit est très courante lors de la modification d’une étape de procédé, changement d’une matière première, scale up, transferts de technologie/procédé dans un nouveau bâtiment ou sur un nouveau site…
Comme précisé en introduction, il existe pour les produits biologiques la guideline ICHQ5E : “note for guidance biotechnological/biological products subjected to changes in their manufacturing process” dont l’objectif est de donner les lignes directrices et principes permettant de démontrer la comparabilité de la qualité produit dans le cadre de modifications de procédés. Cette guideline permet de comprendre l’intérêt et la force de ces études car elles vont permettre de démontrer que l’efficacité, la pureté et l’innocuité du produit, au travers d’attributs qualité du produit (profil de control standard et caractéristiques additionnelles), ne sont pas impactées par la modification. Elles doivent être suffisamment puissantes pour pouvoir justifier de ne pas réaliser d’études cliniques.
Parfois, la modification peut avoir comme objectif d’améliorer la qualité du produit comme par exemple sa pureté et l’étude de comparaison devra alors reposer sur une analyse statistique adaptée.
Contrairement aux biosimilaires, dans le cas des modifications de procédés commercialisés, le critère d’acceptabilité de la référence peut être définit sur un nombre d’échantillons important puisque l’historique de production pour les attributs qualité standards pourra être utilisé. Ceci permettra d’appréhender les sources de variabilité procédé et les taux d’excursions. Par contre, le nombre de lot post modification restera toujours limité. Ainsi, la difficulté de ces études portera sur le choix de l’outil statistique adapté permettant de comparer un critère basé sur un grand nombre de lots de référence à des données obtenues sur un faible nombre de lots.
Il est important de souligner que le document rappelle bien que la comparabilité doit se faire sur plusieurs caractéristiques et que l’approche statistique doit être adaptée à chaque type de caractéristique.
1.2. Les différentes approches statistiques et leurs limites
1.2-1. Choix des caractéristiques à comparer et objectifs
Pour chaque attribut qualité sélectionné, l’objectif est de comparer les résultats des 2 procédés de fabrication.
L’approche statistique doit être fonction du type de donnée : continue ou discrète.
Pour les données continues une des options est de comparer la moyenne des distributions qui peut être complétée par la comparaison de l’écart ou de la variance des distributions.
D’une manière plus générale, la comparaison de données est la plupart du temps basée sur les options suivantes :
- Conformité à des limites de spécifications
Cette approche permet de comparer chaque caractéristique d’un lot à une spécification prédéfinie qui peut être unilatérale ou bilatérale. Dans ce cas, il est important de vérifier comment et quand ces intervalles ont été définis et de s’assurer de leur pertinence par rapport à l’objectif de l’étude mais aussi de s’assurer que le procédé en place au moment de la définition des spécifications est toujours représentatif du procédé actuel qui servira de référence.
- Comparaison de non infériorité
Dans ce cas, on compare deux séries de lots avec comme objectif de démontrer que l’un des 2 procédés ne génère pas un produit de qualité inférieure à l’autre mesuré par des caractéristiques produits appropriées.
En statistique cela correspond à un test statistique unilatéral. Une des approches classiques est de faire une investigation de non infériorité via une comparaison des données à un intervalle unilatéral dérivé d’un échantillonnage réel à un intervalle défini à priori.
- Comparaison à un intervalle bilatéral de similarité / équivalence
Dans ce cas, on recherche plutôt à démontrer que la qualité du produit des 2 procédés est équivalente c’est-à-dire que les deux procédés génèrent des produits de qualité équivalente.
Une des approches est de définir un intervalle de confiance bilatéral et de comparer cet intervalle à une marge d’équivalence prédéfinie.
Cette approche implique l’hypothèse de procédés reproductibles.
1.2-2. Comprendre les sources de variabilité des données qualité
Au travers de ce paragraphe, les autorités de santé ont souhaité insister sur l’importance de prendre en compte le fait que les études de comparaison vont être réalisées à partir de données qualité produit provenant de différents lots et que de ce fait la variabilité du procédé et des méthodes analytiques va entrer en jeu dans la comparabilité. Il est donc important au préalable d’une étude de bien comprendre le procédé et sa variabilité mais également de connaitre la variabilité des méthodes analytiques. Il est donc conseillé d’avoir réduit au maximum les sources de variabilité assignables afin de ne garder que les sources de variabilité intrinsèques du procédé.
Différentes sources de variabilité ont été listées à titre d’exemple sans être revendiquées exhaustives :
- Variabilité inter lots
- Variabilité intra lots
- Variabilité intra échantillons
- Variabilité analytique
Ce point est important car plus le procédé de référence sera variable, plus le critère de comparabilité sera étendu et donc plus la pertinence de l’étude sera réduite. La taille de l’échantillon sera également dépendante de la variabilité globale.
1.2-3. Randomisation des échantillons / approche expérimentale
Comme cela a déjà été abordé plusieurs fois, les données à analyser doivent être représentatives du procédé en évaluer. L’idéal serait d’avoir une approche d’échantillonnage par randomisation. Mais cette approche implique que chaque unité doit avoir la même chance d’être sélectionnée. Dans le cadre d’attributs qualité produit, cette approche n’est pas vraiment applicable puisqu’en général le nombre de lots post modification est limité et que les lots sont fabriqués consécutivement.
Dans ce cas, encore une fois, il est important de connaitre la variabilité du procédé de manière à définir un plan d’échantillonnage représentatif et éviter un échantillonnage trop restrictif ne permettant pas de faire une analyse comparative pertinente. Si le plan d’échantillonnage n’est pas représentatif, alors quel que soit le modèle statistique mis en œuvre, l’interprétation en découlant ne sera pas robuste.
Dans certains cas, un échantillonnage pseudo-randomisé peut être mis en place afin de choisir délibérément des lots représentatifs de certaines conditions.
1.2-4. Les moyens de mesure pour décrire les différences entre deux procédés
Une fois que les paramètres d’intérêt ont été sélectionnés, l’étape suivante est d’identifier les moyens de mesure permettant de décrire les différences entre les paramètres pour les deux distributions de données. Par exemple, pour une analyse comparative de moyennes, le moyen de mesure correspond à la différence des moyennes ou le ratio des moyennes.
La définition de ces moyens de mesure permettant de décrire la différence entre deux distributions de données inconnues revient à calculer la différence existant entre ces deux distributions et donc de simplifier l’analyse.
1.2-5. Intervalles statistiques intégrant la quantification des incertitudes
Le calcul de certains intervalles statistiques peut permettre dans certains cas de quantifier l’incertitude liée au fait qu’une conclusion va être faite sur la base d’un échantillonnage et quelle est applicable à l’ensemble des lots qui seront fabriqués avec ce procédé.
De manière à utiliser au maximum les propriétés des intervalles statistiques dans la définition des critères de comparabilité, il est essentiel que l’objectif de la comparaison ainsi que les moyens de mesure permettant de caractériser les différences soient choisis consciencieusement.
1.2-5-1 approches de comparabilité basées sur les intervalles communément rencontrés
Il est rappelé qu’une distinction claire doit être faite entre la quantification des incertitudes utilisant les intervalles statistiques et la définition des critères de comparabilité.
En pratique, les intervalles ou critères de comparabilité sont fréquemment basés sur un intervalle statistique, comme par exemple : min/max ou intervalle de tolérance calculé pour une caractéristique du produit de référence.
- Approche Min/Max
L’approche Min/Max décrit l’étendue des données pour un échantillonnage mais ne prend pas en compte l’incertitude liée à la distribution des données.
L’approche Min/Max est plutôt conseillée pour comparer des attributs qualité produits de deux séries de lots avant et après modification. L’analyse va donc consister à vérifier que pour chaque attribut qualité produit sélectionné, la valeur min et la valeur max obtenue sur l’ensemble des lots post modification est bien contenue dans la l’encadrement Min/Max définit sur un échantillonnage représentatif du procédé de référence avant modification.
Cette approche a ses limites et est peu discriminante car plus le nombre de lot post modification est faible plus les chances de rentrer dans l’intervalle Min/max de référence sont importantes et donc de revendiquer une fausse comparabilité.
- Intervalle de tolérance & approches x-sigma
Un intervalle de tolérance est généralement calculé pour estimer l’étendue des données pour une proportion (p) de la population couverte avec un degré de confiance (c) de x%.
Alors que l’intervalle de tolérance est conceptuellement approprié pour décrire le niveau d’incertitude lié à une distribution de données inconnue, son utilisation requière une attention particulière :
– Vérification de la normalité de la distribution des données.
– Le choix des valeurs des paramètres p et c
Et nécessite un nombre de lot important afin de compenser les limites de ces approches.
1.2-5-2 principes de calcul des intervalles statistiques pour la comparaison des attributs qualité
- Intervalles de prédiction
Les intervalles de prédictions permettent de décrire un intervalle prédictif intégrant des données qui seront générées dans le futur. Ils peuvent être calculés pour une seule future observation ou plusieurs (k) observations.
- Intervalles de confiances
Ils sont le plus souvent utilisés dans le cas des études de non infériorité lors des études cliniques.
1.3. Les spécificités pour les études de comparabilité dans le cadre d’une modification de procédé
La nécessité d’avoir un échantillonnage représentatif des unités produites est un des facteurs limitants. Car même s’il est facile d’avoir un échantillonnage représentatif du procédé avant modification, il est difficile de faire l’analyse avec un échantillonnage représentatif du procédé après modification. Aucun nombre minimum de lot post modification n’est demandé par les autorités. Mais le nombre de lots devra être défini sur la base de la reproductibilité et variabilité du procédé.
Enfin les autorités demandent que les méthodes statistiques et la taille de l’échantillonnage soient justifiées en tenant compte de la variabilité du procédé.
En synthèse, on observe dans ce document que le sujet lié aux études de comparabilités et d’analyses statistiques associées reste encore confus de par sa complexité et du niveau d’expertise requis. En effet, il manque dans ce document, une différenciation entre les analyses statistiques à mettre en œuvre pour les biosimilaires, pour les modifications ou transferts de procédés / méthodes analytiques post approbation et pour les procédés en phase de développement. Dans ces 3 cas, les objectifs et les conditions sont très différentes et les outils statistiques doivent donc être appropriés. C’est pourquoi, les commentaires généraux résumés dans le paragraphe suivant sont principalement orientées sur des définitions plus précises, une réorganisation du document afin de séparer les 3 grands thèmes : biosimilaires, modifications ou transferts de procédés et méthodes post approbation et procédés en phase de développement/études cliniques.
2. Synthèse des commentaires issus du GIC soumis à l’EMA
Les membres du GIC n’ayant pas d’expérience sur les biosimilaires, cette partie du guide n’a pas été commentée. Les commentaires sont donc principalement orientés sur les modifications ou transferts de procédés et méthodes analytiques post approbation.
- Dissocier les 3 grands domaines : modifications post approbation, biosimilaires et développement des génériques.
- Pour les modifications post approbation :
– Prendre en compte les contraintes industrielles car il n’est pas possible d’avoir 10 lots ou plus post modification pour faire la comparaison
– Prendre en compte les sources de variabilité du procédé
– Renforcer l’étude en considérant les données qui seront générées au travers de la vérification en continue du procédé (CPV)
– Ne pas rejeter l’approche des intervalles (prédiction, x-sigma, intervalle de tolérance…
– Ne pas imposer le test d’équivalence applicable surtout pour les études de comparaison réalisées pendant les études cliniques
– Associer poids et taille d’échantillon dans le calcul et la marge d’équivalence - Afin d’aligner les industriels et les autorités, nous avons demandé à ce que les termes similarité, équivalence et comparabilité soient définis de manière précise. Par exemple, la similarité doit être réservée aux études pour les biosimilaires et comparabilité aux autres produits.
- Des statisticiens non spécialisés en analyses statistiques pour études cliniques participent également à ce document afin de ne pas traiter les études de comparabilités post approbation selon une approche étude clinique.
- Pour les études de comparabilité utilisant l’approche des intervalles, la taille de l’échantillon (le nombre de lots disponibles pour l’étude) doit être en cohérence avec l’étude et restreinte aux études de comparabilités utilisés dans le cadre d’un changement de procédé ou méthode post approbation.
- Une partie du guide doit traiter les approches d’équivalence dans le cadre de changements appliqués aux méthodes analytiques.
- La méthode Bayesian doit pouvoir être utilisée pour les petites tailles d’échantillons et un paragraphe spécifique a été recommandé afin de détailler cette méthode avec ses limites et son champ d’application.
- Dans le cas d’étude comparative impliquant peu de lots pour des changements post approbation et ou si l’approche d’équivalence n’est pas applicable, alors l’approche Xsigma renforcé par un suivi complémentaire doit pouvoir être utilisée. Ainsi, l’étape 3 de la validation des procédés dénommée CPV (Continued Process Verification ou vérification en continu des procédés) doit pouvoir être utilisée pour renforcer la démonstration de la comparabilité sur le long terme.
- Pour certains produits comme les vaccins ou produits biologiques manufacturés par campagne, on observe parfois un effet campagne provenant par exemple de la variabilité des matières premières faisant ainsi partie intégrante des causes communes de variabilité du procédé. C’est pourquoi, cette spécificité doit être prise en compte pour les études de comparabilité et adressée dans le document.
- Une méthode non paramétrique ou “Min/Max” peut être utilisée lorsqu’il n’y a pas suffisamment de données ou si la normalité de la distribution n’est pas démontrée.
- Nous avons également suggéré que l’analyse statistique ne doit pas être le seul élément supportant la comparabilité. Des éléments complémentaires tels que les données cliniques doivent venir confirmer les résultats de l’étude statistique et que la comparabilité doit être statuée sur l’ensemble des éléments : données statistiques, données cliniques et l’expertise procédé / produit.
- Contrairement au document proposant que les critères de comparabilité soient définis uniquement en fonction de la variabilité des données qualité du produit, les membres du GIC ont proposé 2 approches pour définir les critères de comparabilité : soit basé sur la connaissance scientifique et l’expertise produit & clinique soit basé sur l’impact potentiel sur la capabilité du procédé afin de prendre en compte la moyenne et la variabilité des résultats.
- Enfin, nous avons suggéré la refonte du document afin de bien dissocier les différentes approches et les 3 grands domaines : changements post approbation, biosimilaires et développement de génériques, selon la structure ci-dessous.
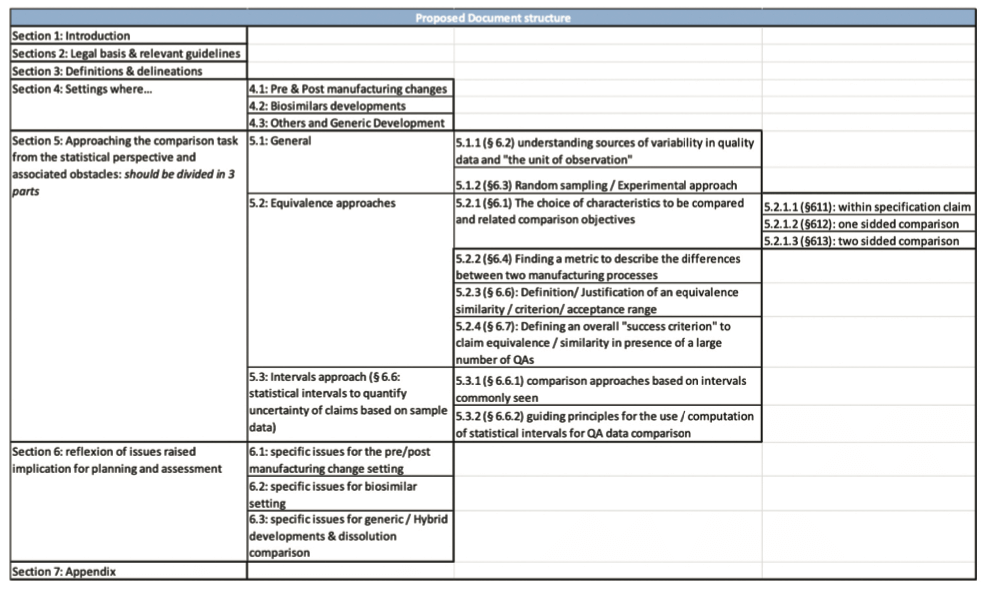
3. Synthèse du workshop organisé par l’EMA à Londres les 3 et 4 mai 2018 suite à la période de consultation publique
Une soixantaine de participants à ce workshop composé d’environ 30 personnes représentant les autorités de santé et 30 personnes issues de l’industrie pharmaceutique ou d’associations pour l’industrie pharmaceutique.
5 sessions ouvertes avec les industriels et pour chaque session, présentation de cas d’études par des industriels ou des représentants des autorités avec en fin de session une partie discussions/échanges.
- Identification de la situation et des challenges :
– discussion sur la nécessité et la pertinence d’appliquer les même critères pour les biosimilaires et pour les modifications post approbation. - Cas d’études sur les modifications post approbation :
– Focus sur la variabilité inhérente des produits et procédés biologiques mais les approches peuvent être communes avec les produits pharmaceutiques standards.
– L’importance de la taille de l’échantillon, de la sélection des lots, la pertinence des caractéristiques produit à comparer, les approches d’intervalles de tolérances (intervalles de prédiction, intervalles de tolérances ou Min/Max) ont été discutées. - Cas d’études sur les biosimilaires
- Caractéristiques fréquemment utilisées pour établir les critères de similarité
- Nouvelles stratégies et méthodes alternatives.
En conclusion du workshop, il a été reconnu que :
- Les différentes approches et domaines doivent être dissociés et séparés dans le document.
- Les approches de définition des critères de similarité pour les biosimilaires doivent être adaptés et plus strictes que dans le cas de changements post approbation.
- Il était difficile de recommander une approche optimale pour définir un critère et que l’alternative possible serait plutôt de focaliser sur la compréhension commune de l’importance d’avoir un critère d’acceptation en place et adapté au cas.
Conclusion
On voit au travers de ce draft que les terminologies et approches doivent être précisées en fonction des domaines et que de nouvelles méthodes statistiques doivent être intégrées.
Néanmoins, il est indéniable que sans les statistiques nous ne pouvons pas faire d’études de comparaison robustes et pertinentes mais tous les intervenants doivent avoir une bonne connaissance et compréhension des procédés, des méthodes statistiques et les besoins doivent être clairement identifiés.
Enfin, comme énoncé en introduction, ce document, s’inscrit parfaitement dans l’évolution des approches de validation des procédés mais aussi de la guideline ICH Q12. La finalité étant de favoriser l’innovation, renforcer la qualité des produits et sécuriser la mise à disposition des médicaments.
Pendant les phases de développement d’un procédé, la connaissance et la compréhension du procédé /produit au travers du “Quality by design” va poser les bases nécessaires à de futures études de comparabilité. La validation du procédé permettra de démontrer un procédé industriel reproductible et sa variabilité intrinsèque. Tout changement intervenant post approbation devra en plus de la validation démontrer la comparabilité du produit et le CPV permettra de renforcer les études de comparabilité. Cette approche globale permettra ainsi de mettre sur le marché des produits surs et efficaces avec des procédés maitrisés et reproductibles.
 | C’est pourquoi A3P a initié 4 GIC sur ces thèmes.
|
Partager l’article


GIC A3P STATISTIQUES
Références
ICH Q5E: Note for guidance on biotechnological/biological products subjected to changes in their manufacturing process (CPMP/ICH/5721/03)
ICH Q8: Pharmaceutical Development (EMA/CHMP/ICH/167068/2004)
ICH Q9: Quality Risk Management (EMA/CHMP/ICH/24235/2006)
ICH Q10: Pharmaceutical quality system (EMA/CHMP/ICH/214732/2007)
ICH Q11: Development and manufacture of drug substances (chemical entities and biotechnological/biological entities, EMA/CHMP/ICH/425213/2011.
ICHQ12: Draft: Technical and regulatory considerations for pharmaceutical product lifecycle management,
Glossaire
GIC : Groupe d’interêt commun
ICH : International Council for Harmonisation
EMA: European Medecines Agency
GMP : Good Manufacturing Practices
CPV : Continued Process Verification
