


Sommaire
- Gene therapy manufacturing comes of age: Commercial-scale manufacturing is imminent. Are gene therapy innovators ready?
- Overcoming obstacles in downstream bioprocessing of AAV based gene therapy products
- Overcoming challenges in the development of lentiviral vector manufacturing platforms
- Are modern scalable bioreactors the Cell Culture Strategy needed for Gene & Cell Therapy success?
- Le pouvoir magnétique des nanoparticules triage magnétique cellulaire dépollution des milieux et nano catalyseurs réutilisables
- Développement d’une nouvelle approche préventive pour réduire les infections microbiennes avec les nanoparticules d’oxydes métalliques
- Surveillance numérique de l’environnement : détecter les défaillances avant qu’elles ne se produisent
- Maximizing Sterility Assurance: Sterile Hold Time Testing for Sterilized Items Used in Parenteral Drug Manufacturing
Maximizar la garantía de esterilidad: evaluación del plazo de validez de los materiales esterilizados utilizados en la fabricación de medicamentos parenterales
El periodo de esterilidad depende del sistema de envoltura esterilizante, de la intervención del operador y del rendimiento del ciclo de esterilización. Tras la esterilización, las piezas y los equipos deben utilizarse dentro de un plazo que se ha comprobado y validado. Este plazo de validez admisible se establece para cada elemento esterilizado y sistema de envoltura esterilizante asociado, y generalmente se confirma mediante una simulación de proceso aséptico (APS, por sus siglas en inglés). Se ha llevado a cabo una evaluación comparativa de los fabricantes de los sectores farmacéutico y biofarmacéutico para determinar si existen prácticas comunes a la hora de establecer los plazos de validez de la esterilidad.

El periodo de esterilidad depende del sistema de envoltura esterilizante, de la intervención del operador y del rendimiento del ciclo de esterilización. Tras la esterilización, las piezas y los equipos deben utilizarse dentro de un plazo que se ha comprobado y validado. Este plazo de validez admisible se establece para cada elemento esterilizado y sistema de envoltura esterilizante asociado, y generalmente se confirma mediante una simulación de proceso aséptico (APS, por sus siglas en inglés). Se ha llevado a cabo una evaluación comparativa de los fabricantes de los sectores farmacéutico y biofarmacéutico para determinar si existen prácticas comunes a la hora de establecer los plazos de validez de la esterilidad.
El plazo de validez establecido se confirma utilizando piezas «caducadas» instaladas en la línea de llenado o en operaciones de llenado aséptico como parte de la APS rutinaria. El uso de piezas esterilizadas al final del plazo de validez en la APS confirma que las piezas permanecen estériles durante el periodo de caducidad. Los estudios del periodo de esterilidad permiten el almacenamiento de las piezas estériles, con lo que se elimina la necesidad de esterilizar los materiales el mismo día del proceso de llenado.
En los materiales que deben ser estériles, las envolturas de esterilización (bolsas y sobres) permiten la penetración del vapor durante la esterilización en autoclave y mantienen una barrera microbiana tras la esterilización. La envoltura permite trasladar piezas y equipos por toda la instalación y dentro de las salas blancas sin poner en peligro el producto crítico al entrar en contacto con superficies. La doble envoltura o el uso de un material de envoltura compatible con la aplicación manual de desinfectantes de superficies son métodos recomendados para reducir el riesgo durante el traslado de material a espacios de clasificación superior. Por estas razones, es esencial definir el sistema de envoltura esterilizante (material de fabricación y método de cierre).
Para probar y confirmar la eficacia del sistema de envoltura esterilizante, se realizaron estudios7 utilizando diversas bolsas, sobres y métodos de cierre durante un periodo de validez de 30 días. Se utilizaron indicadores biológicos para confirmar la esterilización en el momento inicial y se realizaron pruebas de esterilidad durante todo el periodo de validez.
1. Requisitos normativos y directrices de la industria
Numerosas agencias reguladoras (FDA, MHRA, HPRA, ANVISA, EMA) valoran la necesidad de llevar a cabo estudios del periodo de esterilidad para la fabricación de medicamentos parenterales. En cuanto al envasado, se define con más detalle en el recién publicado EudraLex: Volumen 4, Anexo 1 y se aborda también en otros documentos de orientación reglamentaria. También existen directrices de la industria que abordan el uso de piezas estériles dentro de la APS. EudraLex de la Unión Europea: Volumen 4, Anexo 11 8.46: «Siempre que sea posible, los materiales, equipos y componentes deben esterilizarse mediante métodos validados apropiados para el material específico. Después de la esterilización, debe proporcionarse una protección adecuada para evitar la recontaminación. Si los artículos esterilizados no se utilizan inmediatamente después de la esterilización, deben almacenarse en envases debidamente cerrados. También debe establecerse un plazo máximo de esterilidad». 8.48: «Cuando los materiales, equipos, componentes y elementos auxiliares se esterilicen en envases o contenedores sellados, el envase deberá estar cualificado para minimizar el riesgo de contaminación por partículas, microbios, endotoxinas/pirógenos o productos químicos, y para ser compatible con el método de esterilización seleccionado. Debe validarse el proceso de sellado del envase. La validación debe tener en cuenta la integridad del sistema de barrera protectora estéril, así como el plazo máximo de esterilidad antes de la esterilización y la vida útil máxima asignada a los artículos esterilizados. La integridad del sistema de barrera protectora estéril de cada uno de los artículos esterilizados debe comprobarse antes de su uso». Con el mayor nivel de detalle que ofrece el Anexo 1, algunas expectativas están mucho más claras, pero hay otras en las que no se explican los métodos de ejecución. El Anexo 1 ahora dice específicamente que se requieren estudios del periodo de esterilidad, pero no se explica cómo realizar estas pruebas.
USP 12112 : «Equipo en contacto directo con componentes, contenedores, cierres y productos estériles. Los procedimientos utilizados para la limpieza y esterilización de las superficies en contacto directo, incluidos los periodos de esterilidad, limpieza y suciedad, deben validarse para garantizar que no afectan negativamente a los atributos esenciales de calidad del producto, así como para verificar la eficacia del procedimiento de limpieza y que no se produce recontaminación/proliferación microbiana durante el almacenamiento del equipo».
La Asociación de Medicamentos Parenterales (PDA, por sus siglas en inglés) ofrece la siguiente orientación a la industria en el Informe Técnico n.º 22, Simulación de procesos para productos llenados asépticamente5 . En esta sección, la información describe los ejemplos más desfavorables de desarrollo de la APS. 3.2… Otros ejemplos de prácticas más desfavorables pueden ser: Utilización de la sala/equipo en el periodo de tiempo máximo tras la finalización de la higienización/esterilización (periodo de limpieza). En el número de marzo/abril de 2022 de Pharmaceutical Engineering4 se analizan las consideraciones normativas y de APS a la hora de desarrollar la simulación para satisfacer todas las necesidades del proceso. La lista que figura a continuación contiene ejemplos de algunos de los retos más complejos que deben abordarse en el marco de una APS. Página 51: APS Considerations- Worst Case Challenge:… Hold Time Equipment/room clean hold time Equipment sterilization hold time.
2. Ventaja operativa
Las pruebas del periodo de esterilidad garantizan la integridad del envase para proteger la pieza o el equipo estéril. Los envases pueden conservarse en una sala limpia después de la esterilización y pueden estar listos para su uso cuando se necesiten sin tiempo de preparación adicional. El autoclave tiene una alta demanda en la mayoría de las instalaciones y el tiempo de reprocesamiento adicional es costoso; además, la disponibilidad inmediata puede ser limitada. La confirmación de un periodo de validez también permite un menor reprocesamiento de las piezas/equipos esterilizados en autoclave si no se utilizan dentro de la producción el mismo día de la esterilización. Desacoplar el plazo de esterilidad de la APS reduce las posibles variables si se produce un fallo. Debido a que se han estudiado los peores escenarios, la eliminación de cualquier variable innecesaria permite una investigación más ágil y la determinación de la causa raíz.
3. Recalificación
Como se ha mencionado anteriormente, el periodo de esterilidad se confirmaría durante una APS. El uso de material caducado se recalificaría durante estas pruebas. Sin embargo, el propósito de la APS no es únicamente confirmar el plazo de validez de la esterilidad. Dentro de una APS, hay múltiples variables que se evalúan como escenario más desfavorable. Debido a las múltiples variables, determinar la causa raíz de un fallo es difícil. Separar los estudios del periodo de esterilidad y la APS reduce las variables durante la simulación. De no producirse este desacoplamiento, sería difícil señalar las piezas esterilizadas como causa principal en caso de que falle la simulación.
4. Diseño de experimentos
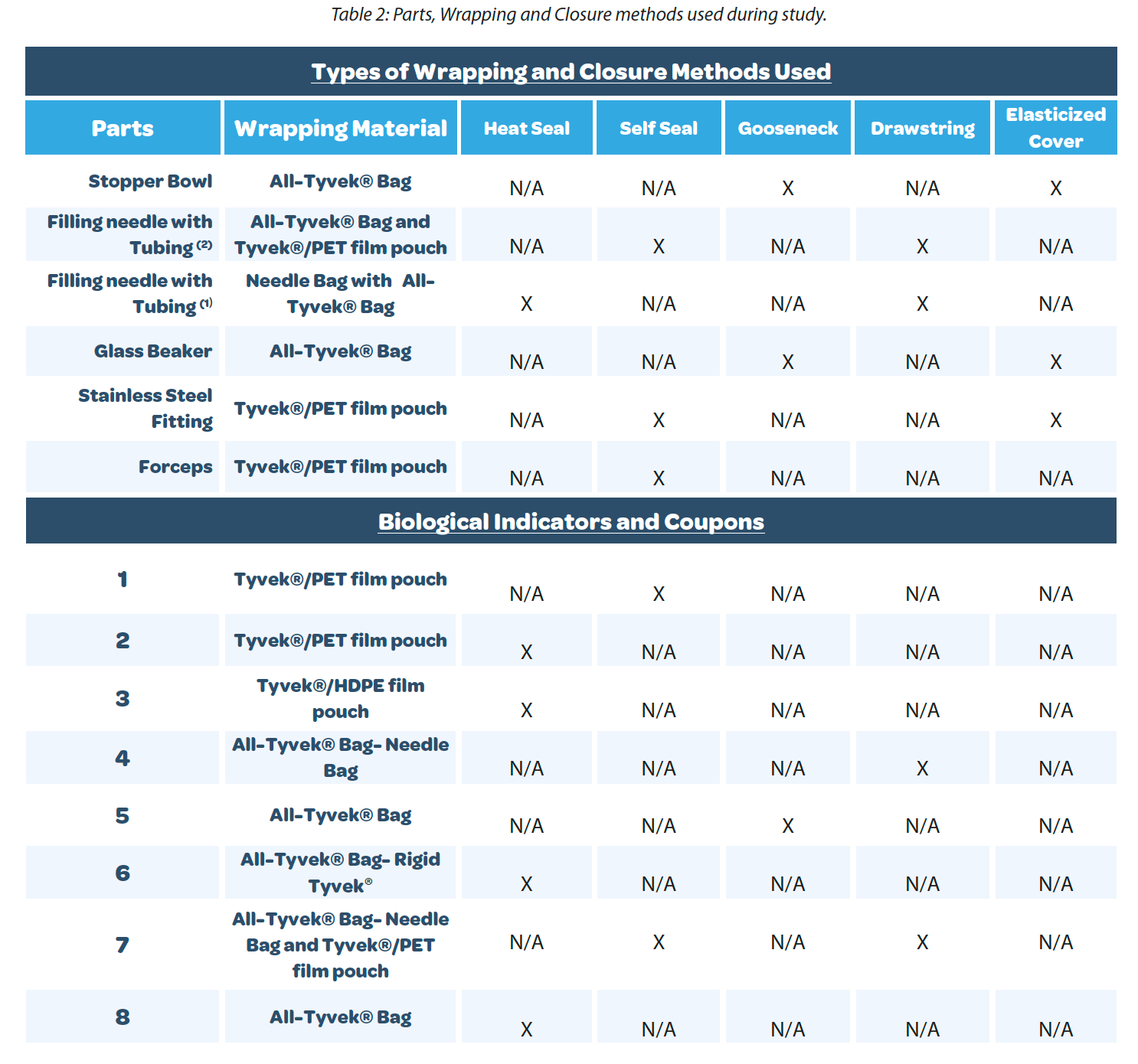
La garantía de esterilidad se maximiza confirmando los periodos de validez de las piezas o equipos utilizados para la fabricación aséptica. Cada usuario final debe validar las configuraciones rutinarias de envoltura utilizando piezas y equipos representativos durante un intervalo de tiempo que se ajuste al proceso individual. Esta envoltura debe ser fácil de reproducir por los empleados para garantizar la coherencia. La consistencia de la envoltura puede ayudar a confirmar que la barrera microbiana está intacta después de cada ciclo de esterilización. El periodo de validez de la esterilidad debe ser confirmado por el usuario final y comprobado durante una APS. Durante un estudio del periodo de validez, hay múltiples variables que es necesario probar para crear un estudio exhaustivo. En el Cuadro 1 figuran algunos ejemplos de la industria biofarmacéutica.
Debe evaluarse el traslado de piezas a través de las instalaciones. Las piezas deben prepararse y envolverse de forma que se facilite la transferencia de material. La forma en que se produce el proceso de transferencia después de la esterilización dentro de una esclusa de manipulación de materiales puede tener un impacto importante en la integridad de la envoltura, que a su vez puede afectar a la barrera microbiana de la envoltura. Como caso ilustrativo, la desinfección de la superficie de la envoltura o la extracción de una capa exterior de la misma deberían ser evaluadas y validadas. A continuación, el proceso de transferencia de material permite colocar las piezas estériles envueltas dentro de la zona de sala blanca controlada. La configuración final de la envoltura después de la transferencia del material a una zona limpia es la envoltura que se prueba para el periodo de validez de la esterilidad.

Las piezas y los equipos elegidos durante el estudio determinarán las configuraciones de envoltura que se probarán. El material de envoltura debe generar pocas partículas para limitar el riesgo para el medicamento. También debe ser compatible con los métodos de transferencia a una zona aséptica. La transferencia suele completarse mediante la aplicación de desinfectantes o alcoholes sobre el elemento envuelto antes de transferirlo de las clasificaciones inferiores a las zonas más críticas. Una vez en las clasificaciones superiores, eliminar la capa externa del material de envoltura, si esta es doble, puede ser un método de transferencia de material más rápido y eficiente. La envoltura de las piezas se decide en función del proceso y de la presentación aséptica durante el uso de la pieza, ya que quitarle la envoltura después de la esterilización es el mayor riesgo de contaminación de la superficie estéril. La facilidad de desenvoltura en el momento del uso minimiza el riesgo de contaminación por parte de los operarios y del entorno. Por ello, el sistema de envoltura debe configurarse para cada proceso y pieza específicos. Los métodos de cierre deben probarse en el marco de un estudio del periodo de esterilidad. Las mejores prácticas de cierre evitan los materiales que puedan generar partículas, como los materiales a base de celulosa («envoltura azul») y la cinta de autoclave dentro de la zona aséptica. Según el Anexo 1, Sección 8.48, «Cuando los materiales, equipos, componentes y elementos auxiliares se esterilicen en envases o contenedores sellados, el envase deberá estar cualificado para minimizar el riesgo de contaminación por partículas, microbios, endotoxinas/pirógenos o productos químicos, y para ser compatible con el método de esterilización seleccionado…». Los métodos de cierre más recomendables permiten extraer las piezas del envase con el mínimo contacto con el personal o con las herramientas que podría dar lugar a contaminación y presentar riesgos para el producto final. Las coberturas elásticas se utilizan como barreras iniciales para las superficies en contacto con el producto, pero no se consideran una barrera microbiana completa. Permiten una protección adicional al instalar piezas y equipos que están en contacto con el producto para limitar la contaminación por manipulación del personal durante la instalación y la puesta en marcha de la línea. El plazo del estudio depende de las restricciones de procesamiento del usuario final y de las piezas. Por ejemplo, para procesos de llenado complicados, puede ser necesario un plazo de 30 días por razones de flexibilidad, mientras que los procesos más sencillos pueden necesitar solo 7 días, ya que las piezas pueden utilizarse rápidamente. Se comprobará la repetibilidad con el mismo plazo de tiempo. Se pueden probar múltiples plazos de tiempo para aumentar la confianza en periodos de esterilidad más largos.
5. Metodología
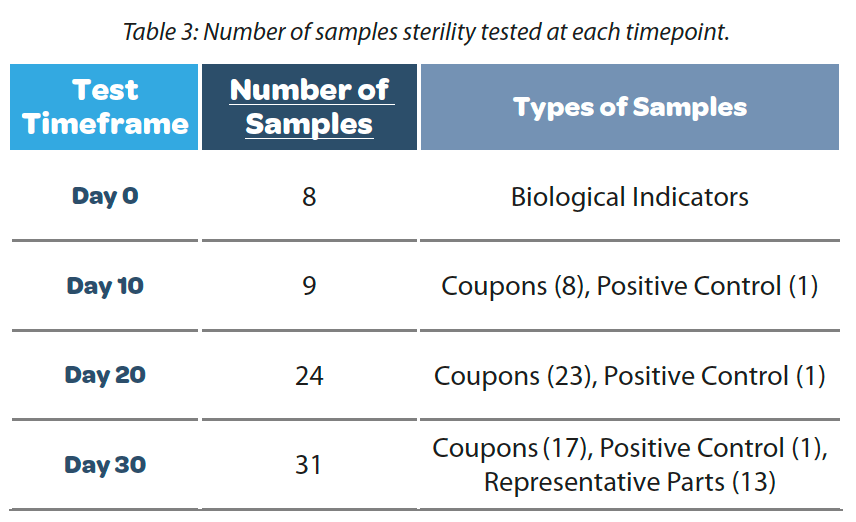
Se realizó un estudio utilizando las piezas, los materiales de envoltura y los métodos de cierre que figuran en el Cuadro 2. Para iniciar el estudio, se eligieron piezas representativas basadas en elementos de uso frecuente en el proceso de llenado/acabado aséptico farmacéutico. También se seleccionaron los elementos más complicados en función de su tamaño, es decir, los más grandes y voluminosos y los pequeños y afilados, así como los difíciles de envolver. Las piezas probadas fueron un recipiente con tapón, accesorios de acero inoxidable, tubos, agujas de llenado, pinzas y vasos de precipitados de vidrio. Estas piezas se envolvieron con las soluciones de envoltura8 recomendadas en el Cuadro 2. Los métodos de cierre, que figuran en el Cuadro 2, se aplicaron en función del material de envoltura utilizado. Todos los cierres probados son las recomendaciones estándar para cada estilo de envoltura utilizado. Además de utilizar piezas representativas, se evaluaron cupones de acero inoxidable y vidrio para minimizar la variabilidad de las pruebas y garantizar la coherencia. El uso de los cupones permitió realizar pruebas de inmersión total en un medio de crecimiento. Se evaluó una duración de 30 días como duración general para esta evaluación, con pruebas de esterilidad realizadas a los 10, 20 y 30 días. Los plazos de tiempo se incrementaron de 10 en 10 días, lo que permitió recopilar más datos para cada configuración de envoltura en caso de fallo en el plazo más largo de 30 días. Los materiales de mayor tamaño y las piezas representativas solo se sometieron a prueba a los 30 días, con resultados satisfactorios. Durante el período de prueba, se colocó un control positivo para cada plazo de tiempo probado en el área de almacenamiento no controlada con las piezas envueltas y esterilizadas. Se envolvieron cupones de acero inoxidable en ocho configuraciones (Tabla 2) con diferentes métodos de cierre y se sumergieron en caldo de soja tríptica (TSB, por sus siglas en inglés) durante catorce días para probar la esterilidad según USP 43 < 71>6. En el Cuadro 3 se indica el número de muestras analizadas en cada periodo.
6. Resultados
Véase el resumen de resultados en el Cuadro 4. Los resultados recopilados durante este estudio mostraron los datos esperados. Los ocho indicadores biológicos dieron negativo en crecimiento, lo que confirma que el ciclo de esterilización realizado en las piezas fue un éxito. Las muestras positivas para cada uno de los plazos de tiempo fueron los controles positivos. La segunda muestra positiva en el día 30 correspondía a una muestra que estaba en una pequeña bolsa All-Tyvek sin cierre sellado, solo un cordón de ajuste. Esto demuestra la importancia de un cierre sellado para garantizar que la barrera microbiana esté intacta. Estos datos confirman que es necesario un cierre de cuello de cisne (recorrido sinuoso) para que una bolsa con cordón mantenga la esterilidad del contenido.

Conclusión
La realización de un estudio del periodo de esterilidad maximiza la garantía de esterilidad de sus piezas y equipos, así como del producto farmacéutico acabado. También ayuda a reducir el reprocesamiento de piezas y equipos estériles. Esta evaluación permite a los usuarios finales almacenar piezas y equipos esterilizados dentro de la zona limpia para limitar la cantidad de esterilización necesaria el día del procesamiento. Las piezas probadas y las configuraciones de envoltura deben imitar las prácticas habituales de envoltura. Si está justificado, es posible reducir el número de piezas que necesitan ser evaluadas con un método basado en el riesgo. Las pruebas aquí resumidas se refieren a un periodo de esterilidad de 30 días utilizando sobres, coberturas y bolsas Tyvek®, y pueden utilizarse para ayudar al usuario final en el diseño de un protocolo de estudio, así como para proporcionar confianza en el plazo de integridad del envase. El cumplimiento de la normativa industrial es responsabilidad del usuario final/fabricante, en función de sus circunstancias particulares.
Partager l’article


