


Summary
- Three steps to contamination control when utilising single use equipment
- Automatic Particle Measurement in Hot Air Sterilizing Tunnels
- Hydrogen Peroxide Sterilization and Isolator Connections through a Magnetic Driven Door For Innovative Lay-outs in Aseptic Processes
- Fast X-Ray Tomography Techniques : ready for new Pharmaceutical Applications ?
Three steps to contamination control when utilising single use equipment
Over the last decade, single-use systems are gaining greater acceptance in biopharmaceutical manufacturing as companies increase the diversity of products they manufacture and redesign processes based on shorter, multi-product runs. Single-use technology has become recognised as increasing important for the drug manufacturers who want to preserve space, increase flexibility, and save money during their manufacturing activities.

Single-use, or disposable systems, are being used to replace expensive, stainless steel bioprocessing equipment such as filter housings, hold tanks and transfer lines, with disposable filters, bioprocessing bags and tubing sets. Single-use systems offer many benefits over stainless steel equipment, including faster implementation, reduced validation requirements, cost savings and lower capital investment. Finally, single-use product can improve overall production yields as part of a manufacturer’s integrated contamination control strategy.
There are a number of potential sources of common contamination in bioprocessing; cross contamination, microbial contamination and biologic contamination of the process facility. These contamination types can be addressed through the use of single-use systems.
The potential for cross contamination occurs when process equipment is used to produce more than one drug product. Unwanted contamination may reduce production yields by requiring additional purification steps, or may result in potentially fatal treatments.
To prevent cross contamination in stainless steel processing equipment, manufacturers must develop and validate clean-in-place (CIP) procedures that remove all residual proteins between processes. CIP cycles typically use large amounts of caustics, acids water-for- injection (WFI) and require regular testing to verify effectiveness. Even simple equipment changes such as adding a new valve may require partial or complete CIP revalidation, increasing time and labour costs.
Single-use systems can reduce or eliminate the need for CIP by starting with virgin polymers that meet USP Class VI biocompatibility and extractables requirements.
However, extractables is always a source of a concern when drug suppliers are implementing the single use systems for the manufacture of drug products. A recent survey by Biopharmaceutical international detailed that out of a range of ten areas, drug suppliers voted lack of extractables and leachables data on single use system components as their number one concern(1). Extractables are chemical compounds that have the potential to migrate from a product contact material into a solvent under worst case or laboratory conditions. It can provide detailed information for drug manufacturers to make an assessment of the polymeric materials in contact with the drug product or drug substance. Although extractables data is not always representative of all that could be obtained from a leachable study, it does provide an indicator what could be potentially leaching into the drug substance. The element of risk as a result of potentially unsafe extractables is a real one. Leachables are defined as the compounds which migrate from the components in the single use system into a biopharmaceutical under the conditions of use. Leachables can have an adverse effect on the drug product in terms of quality, safety and efficacy. Examples where leachables have been found to impact the quality of the drug product include the reaction of therapeutic proteins with acrylic acid which leached from prefilled syringes(2). Another recent example is how the presence of degradation product of a secondary antioxidant, Irgafos 168 was found to be detrimental to cell growth. The degradation product, bis (2,4-di terbutylphenyl)phosphate (bDtBPP) was found to be produced as a result of gamma irradiation of polyethylene films of the bioreactors used to cultivate cells(3). In the example of the bioreactor film leachate although the compound had an impact on the cell viability, recent studies have shown that these breakdown products when leached upstream do not persist in downstream processing after filtration and other purification steps(4). The impact of leachables can be as far reaching as to affect the patient directly. The example of a leachable causing an auto immune response for patients administering a therapeutic used to treat a blood disorder is a one such case(5).
As described above leachables from single use systems could have an impact on the drug product quality and efficacy. Additionally, assessing the risk posed by leachables, which are potentially present in final drug product, is a regulatory requirement of biopharmaceutical manufacturers. Key to understanding the risk posed by leachables from single-use components is data based on extractables test methodology, such as those described by BPOG(6) and soon to be published USP, which enables end users to make informed decisions on patient safety. The extractables data will be able to serve as predicators of what the potential leachates could be in the drug product and could be used as information to perform a risk assessment.
Many different types of polymers could be present in a single use system like a tubing set. This can include polypropylene used in the connectors, polydimethylsiloxane used in tubing and gaskets as well as nylon used in the clamps. The case for understanding the identity of secondary contact components has become more prevalent in recent years. In the wider industry there have been examples of leachates from label adhesive on biocontainer surfaces being found in the drug substances.

A recent study of a tubing set consisting of silicone tubing, silicone gaskets, nylon clamps and polypropylene connectors have found a number of extractables . These extractables are detailed in the paragraphs below.
The extractables found are mostly of cyclosiloxanes from the silicone tubing and gaskets. Cyclosiloxanes are small oligomers of polydimethoxysiloxane (PDMS). The most commonly found siloxanes are hexamethylcyclotrisiloxane (D3) octamethylcyclotetrasiloxane (D4), decamethylcyclopentasiloxane (D5) and dodecamethylcyclohexasiloxane (D6). Other larger congeners can also be found and were identified broadly as siloxane related.
The extractables from the polypropylene hose barb connectors are found to be a range of different compounds. Primarily, these found to be breakdown products the antioxidants used to protect the polymer from oxidative reactions during gamma irradiation or other high energy processes which can promote cleavage of the polypropylene carbon – carbon and carbon – hydrogen bonds. Other compounds found from polypropylene connectors extracts included isomers of dimethylbenzaldehyde, long chain alcohols and fatty acids based on a C10 structure and larger.
The extractables from the nylon clamps are associated with the breakdown products of the polymer. The compounds like caprolactam which is the monomeric unit from Nylon 6,6 and 1,4 butanediol a potential reduction product from the presence of diacid used in the manufacturing of the polymer were readily identified from the clamp extracts.
The number of semi volatiles compounds and their estimated concentrations from the different components are shown in Graph 1.
The silicone gaskets contained the most number of extractables, but these were in low concentrations. The average concentration of extractables per component is detailed in Table and represented in Graph 2.
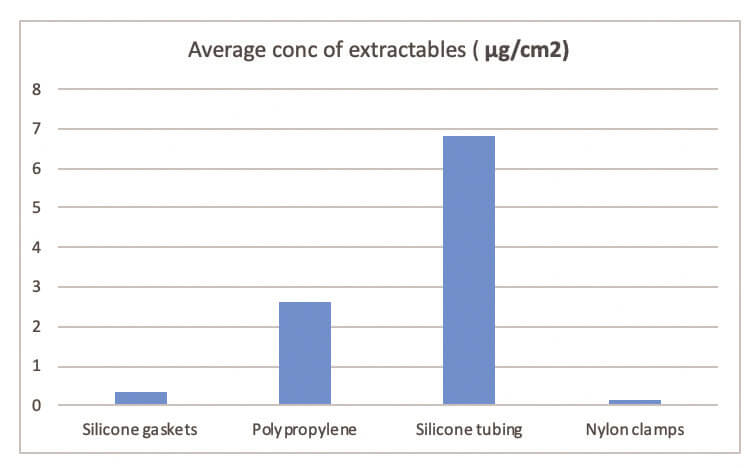
As it can be seen that the non fluid contact material, nylon clamps has the lowest average concentration of extractables. From the fluid path components, the highest average concentration of extractables was from the silicone tubing. This is likely to be due to the higher surface area of the tubing tested.

Discussion
Once the extractables have been identified and quantified, determining whether extractables are of a safety concern can pose a challenge. There are a number of approaches to determining whether chemical compounds present a toxicological impact on human health. The approaches to be discussed below are used in particular to evaluate the mutagenic and genotoxic potential of compounds.
One approach is assessing compounds using the Cramer classification. The Cramer classification was developed for the food industry in 1970s and categorises a group of compounds by three classes. Class 1 being compounds of low or no toxicity, Class 2 medium toxicity and Class 3 high toxicity. A particular drawback of this approach is, it is limited to the compounds that formed the original study. Compounds with unknown toxicity data are given a Class 3 designation in order to provide a worst case estimate(7).
Another methodology that could be used when determining the toxicity of chemicals is to compare the chemical structure of an unknown chemical with that of a compound with known toxicity. There are several ‘in silico’ or computer based packages that can predict the toxicity of a compound based on the similarities of its chemical structure with a known toxic compound.
Popular packages include DEREK, leadscope and multicase can be used as predicative tools of chemical toxicity using a quantitative structure activity relationships (QSAR)(8).
Further commentary about toxicological impact of compounds on human health are detailed in guidelines for potential genotoxic compounds published by ICH and EMEA. They describe an approach to risk assessment which involves the concept of the Threshold of Toxicological Concern (TTC). TTC is a pragmatic approach in risk assessment. It is applicable when there is no specific risk to human health when the exposure is below the threshold level. Risk assessment is the scientific process that characterises the magnitude of risk that chemicals or biologics pose to human and environmental health. TTC may be applied to evaluate materials for their potential toxicity when exposure is very low. In addition, the EMEA guideline proposed a toxicological concern (TTC) threshold value of 1.5 μg/day intake of a genotoxic impurity which is considered to be associated with an acceptable risk (excess cancer risk of <1 in 100,000 over a lifetime) in most pharmaceuticals(9). Based on the TTC value, a permitted level of an active substance can be calculated concerning the expected daily dose. A further approach when looking at toxicity is to determine if there have been any human or animal studies performed to evaluate the risk to human health in the presence of chemicals. This information will provide permitted daily exposure limits. Given the different approaches that could be taken to risk assess and evaluate the toxicological effect of chemicals, combining all to provide a good understanding of risk of the extractables from single use system components could be complex.
Other than extractables data, manufacturers utilising single use equipment may have concerns about contamination sources that could affect their drug products. Microbial contamination is one of the greatest threats to bioprocesses. Once microbes – such as bacteria, yeast and fungi – enter the nutrient-rich bioprocess environment, they quickly overwhelm mammalian cell cultures, utilizing all available nutrients and secreting unwanted and harmful proteins or endotoxins. A bacterial contamination can destroy product valued at millions of dollars in a matter of hours. Endotoxin and bioburden are aspects to be evaluated in final drug products according to a number of regulatory guidance(10).
Endotoxins are fragments of dead Gram negative bacteria that are toxic to the human body which can cause fever or fatal reactions. A frequent source of endotoxins and bioburden is water, which can carry Gram negative bacteria. It is therefore important to examine process water usage. Endotoxin can come from water further back down the supply chain – for example from extrusion, grinding, milling or cleaning processes.
The fact that endotoxins are produced by the death of bacteria means, paradoxically, that sterilisation can sometimes liberate more endotoxins. Prevention is the preferred method of controlling them, particularly since the removal treatments require high temperature and strong chemicals, which are not suitable for most plastic materials.
Limulus Amebocyte Lysate (LAL) is the test reagent. It reacts specifically with bacterial endotoxin, highlighting its presence. For a product to carry any kind of claim, such as “endotoxin free” or “non-pyrogenic”, tests must be carried out to prove this.
Bacterial endotoxin content was tested using the LAL gel clot method described in USP Bacterial Endotoxins Test. Endotoxin was extracted from the tubing with pyrogen-free water and the extract was exposed to the quantity of LAL reagent sensitive to 0.125 EU/mL endotoxin. If the extract does not cause clotting, the test article contains less than 0.125 EU/mL of endotoxins and the test article passes the test. Manufacturers of single use components could provide data on endotoxin evaluations that have been completed on their products, provide end users with confidence and aid them in their contamination controls.
A further source of contamination for users of single use equipment is the presence of particulates. These particulates may as a result of manufacturing processes, when the components are assembled. Most single use providers have standard operating procedures and controlled manufacturing processes to ensure that the generation of particulates is reduced. The concern about particulates arise from due to regulatory guidance which encourages the evaluation of particulates in final drug products. The test methodology for this is described in USP 788 which details the criteria for sub visible particulates in injectable drug products. There are also chapters relating to particulates in opthalamic preparations, USP 789 and visible particulates, USP 790.
Particulate matter is generally defined as mobile, randomly sourced, extraneous substances (other than gas bubbles) that cannot always be quantified; it is typically classified as the following based on its origin:
• Inherent: particulates derived from the product itself, such as protein agglomerates
• Intrinsic: particulates generated from the production process and primary packaging, such as glass vials
• Extrinsic: particulates originating from outside the process, such as garment fibres.
Particulate matter testing forms part of the USP to ensure that unintended and non- therapeutic particulates in solution dosage forms do not exceed established limits. The size of the particulate matter is a critical factor when considering the potential risk to patients. Particulate matter is typically classed as being visible (>100 μm) or subvisible (1–100 μm). The number of less than 10 μm particles can be used as a quantitative impurity test, since they present the highest risk because they are respirable. The ≥ 10 μm and ≥ 25 μm particles also can be shown to have impurity-limits compliance, because they present less risk and are monitored more from a product quality standpoint than a safety risk. With USP 788 there are specific limits for 10 micron and less and 25 micron and less particulates depending on the volume of the injectable.
Conclusion
Concentration of extractables from single use system components tends, to be relatively low. Furthermore depending on where the single use system components are used in the biomanufacturing process, potential leachables could be diluted by large production volumes, thereby ensuring that the levels of leachables in the final drug product is low enough to not present a safety concern to the patient. Contamination controls in the areas of particulates and endotoxin allow end users to assess the contribution of these attributes to their final drug products. Suppliers of single use equipment can provide details of regular assessment of their manufacturing controls used for continued verification of the safety of their products.
Share article


Dr. Sade MOKUOLO – WATSON-MARLOW
Dr. Sade Mokuolu is Global Product Compliance Manager at Watson Marlow Fluid Technologies Group (WMFTG). Prior to joining Watson Marlow, she was employed at Pall providing technical guidance on E & L SUS studies and has presented at international conferences on E & L testing on behalf of the BPSA, as well as delivered training to Australian and European regulators. Additionally, she has extensive pharmaceutical manufacturing experience gained whilst employed by SAFC and Aesica Pharma as a Senior Process, Research and Development chemist.
References
(1) Bioplan associates, 12th annual report and survey of biopharmaceutical manufacturing, April 2015
(2) Norwood et al, – Journal of chromatography, 2009, 11-12
(3) Lee et al, Tungsten leaching from prefilled syringes and impact on protein aggregation, PDA poster,2009
(4) C. Ta et al, Journal of Chromatography A, 1492, 2017
(5) Hammond et al, Biotechnology Progress 30 , 2014, 332-337
(6) BPOG extractables protocol, Ding et al, ISPE, 2014, 34,
(7) Cramer, J.Cosmet. Toxicol.16, 255
(8) www.qsartoolbox.org
(9) ICH M7, 2017 & EMEA, CHMP, 2006 – Guideline on limits of genotoxic impurities
(10) USP 85, Bacterial Endotoxin test
(11) USP <787> Subvisible Particulate Matter in Therapeutic Protein Injection
(12) USP <788> Particulate Matter in Injections
(13) USP <789> Particulate Matter in Ophthalmic Solutions
(14) USP <790> Visible Particulates in Injection
