


Summary
- Limit between GMP Part I/ Part II : Applicability to biological products
- Moving One Unit Operation At a Time Toward Continuous Biomanufacturing
- « Close Collaboration Maximizes Value of Engineered Solutions and Saves Time in Start-Up »
- Improving Single Use Bioreactor Design and Process Development. New Research Towards Intensifying Seed- Train & Scale-up Methods Using 5:1 Turn- Down
- Qualification approach for the validation of real-word shipping in single-use systems
- Expansion of Human Bone Marrow-Derived Mesenchymal Stem Cells in BioBLU 0.3c Single-Use Bioreactors
- Single Use & Stainless Steel: complementarity or fight?
- Low Endotoxin Recovery (LER) is today one of authorities serious concerns regarding pyrogen testing
Following to questions raised by French Biotech industry on GMP part I and II cleavage proposed by inspectors regarding biological production process, the objective of this article is to propose a clarification in the particular case of biological active ingredients when formulation is performed during Drug Substance manufacturing instead of during Drug Product manufacturing.

This apply fully to recombinant proteins and monoclonal antibodies and case by case to other biotechnology manufacturing processes and innovative therapeutic (vectors of gene therapy and vaccines,…)
Problematic
Context
During the production of a chemical ingredient, the stages of synthesis of the active substance (DS) and the transformation into pharmaceutical form of the finished product (DP) are separated in time and often geographically. During the production of a biological substance, and particularly during its purification, it is essential in the majority of the cases to add immediately agents allowing its stabilization. Indeed, a biological substance not being stable would risk denaturation and so to lose its native conformation or to undergo degradations very rapidly. The degradation of the protein of interest or its denaturation would lead to a loss of its therapeutic efficiency and/or to a risk of immunogenicity.
The problematic discussed in this document is intended to propose a clarification for the regulatory referential applicability concerning:
• manufacturing and packaging in bulk of the biological active substance,
• manufacturing of the biological medicine from the bulk of the active substance until its final packaging.
Therefore, modalities of application of this referential could have consequences on the regulatory status in France of the manufacturing site.
Manufacturing process of a biological product
The particularity of a biological product manufacturing process is the need to perform a formulation of the active substance during the stages of purification or to obtain a stabilized bulk which can be stored.
So, it is most of the time a formulated active substance which is released by the production site of the active substance.
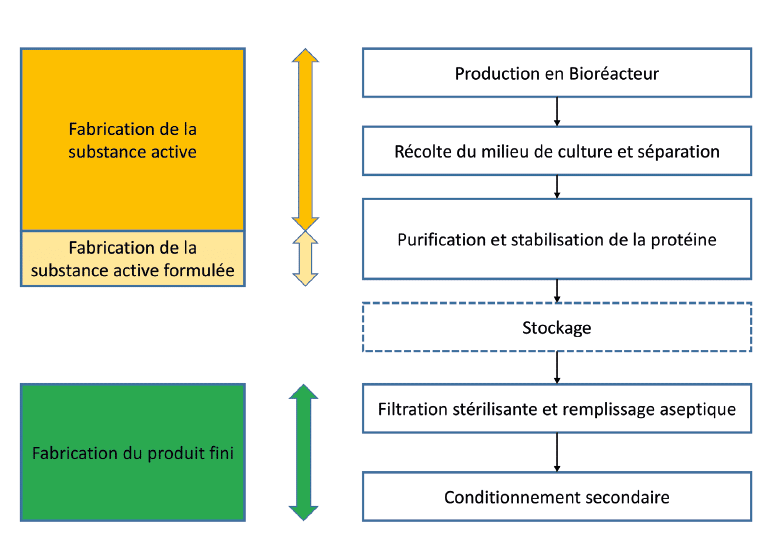
Biological active substances formulation
The formulation of a biological active substance, such as a monoclonal antibody for example, consists mainly of its stabilization.
The biological substance being vulnerable to external factors, thus the formulation step is realized directly during its purification, by the inclusion of stabilizing agents in the purification buffers.
The addition of components at the end of purification is necessary to stabilize the antibody or the therapeutic protein shortly after its extraction.
It is also a question of reducing at the minimum level the freezing/thawing and storage steps of the product, to decrease the risk of denaturation of the product, and to limit the stages of “Intermediate product”.
Therefore, it is frequent that this stabilization during the process is the last stage of manufacturing of the product before the sterilizing filtration and the distribution of the medicine.
The product which is then released by the manufacturer is the formulated active substance, and not a pure active substance, as this is the case for chemicals. This current situation raises the question of the level of segmentation between GMP Part I and II.
Discussion
GMP Part I and II
In this context of biological products, it is necessary to re-specify the scope of applicability of each part of the GMP referential, knowing that annex 2 of the GMP covers the whole manufacturing process of these biological medicines and the level of GMP requirements associated.
In France, the regulations clearly states that the application of part I involves the notion of pharmaceutical establishment manufacturer.
The application of the GMP part II in more or less early stages of production of the active substance does not imply to be pharmaceutical establishment.
The recommendation of the Leem Bioproduction group is summarized on the following diagram which compares the manufacturing stages of a biological medicine to those of a chemical medicine and proposes the level of cleavage between the two parts of GMP for the two types of product.
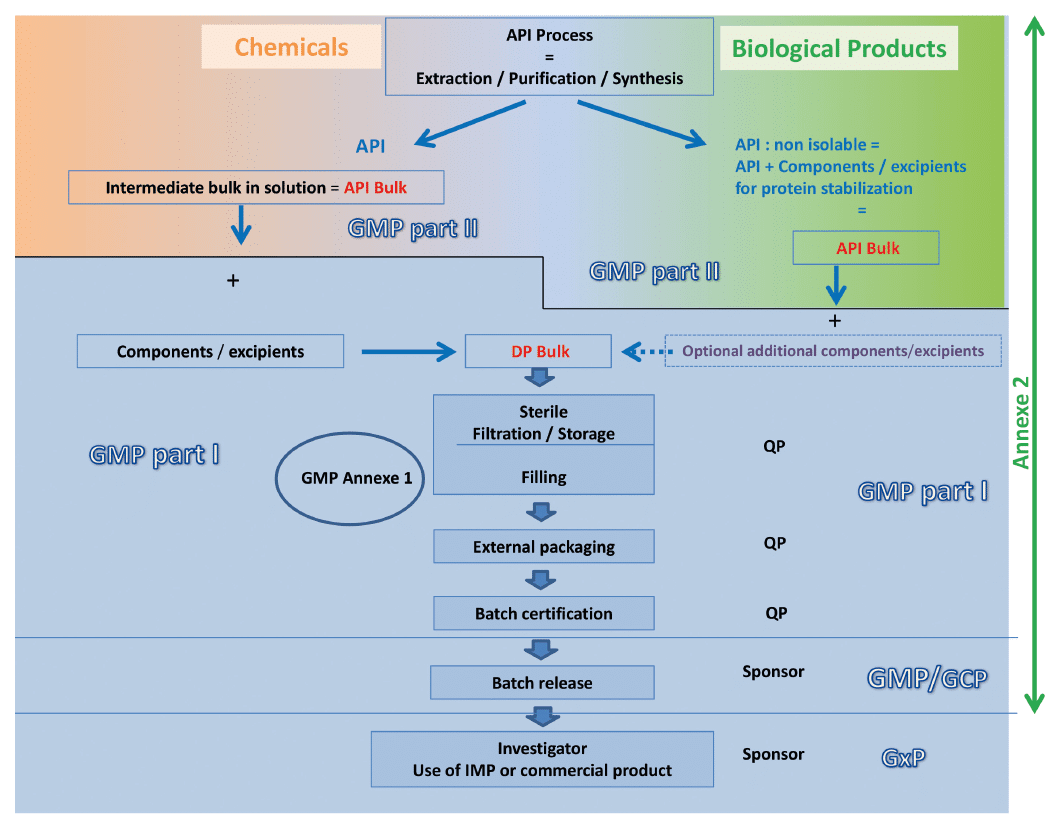
Our reading of the GMP is based on the following points:
• Excipients are necessary for the stabilization of the protein
• Excipients are generally inseparable of the active substance manufacturing process
• The status of the bulk active substance is claimed “low bioburden” rather than “sterile” (even if a filtration 0.2 µ is performed) because the sterilizing filtration is realized afterward, in the stages of manufacturing of the finished product
• It is also possible to make the difference between the formulated active substance and the manufacturing of the Drug Product by the presence of a storage step in the process, the storage step giving rise to stability testing and validation of the sterile storage
• The manufacturing of the active substance until its conditioning is described in the “Substance” part of the regulatory file (submitted to the GMP part II) while the manufacturing of the medicine (Finished product / Drug Product) is described in the “Product” part (submitted to the GMP part I). This allows to ensure the coherence between the submission file and the inspection
So, as mentioned in the GMP Annex 2, the application of the GMP Part I concerns the manufacturing process of the finished product, from the sterilizing filtration or from a storage step, that is the aseptic filling in its entirety (filtration + distribution + integrity of the units to be injected).
When the manufacturing process of a biological product does not contain a sterilization step, the GMP part I applies to upstream stages defined on a case by case basis. The Manufacturer of formulated active substance in the case of biological products, is then not in the obligation to have the status of pharmaceutical establishment.
Remark on the controls of the biological entity : taking into account of the specificity of the biological product manufacturing steps, the active substance is generally of the same composition as the finished product. This can justify that the specific analytical controls of the finished product, requiring particular equipment and expertise, can be performed on the production site of the active substance without being pharmaceutical establishment but as a subcontractor of the pharmaceutical establishment releasing the medicine.
Conclusion
The French regulations require that the manufacturers of pharmaceutical active ingredient comply with the request of authorization for activity of active substance manufacturing with the ANSM for the production of bulk products, without taking the status of pharmaceutical establishment.
In other words, it is not necessary to be pharmaceutical Establishment to manufacture a formulated biological active substance and pharmaceutical companies confirm that it is coherent to claim the application of the GMP part I only from the stage of sterilizing filtration or aseptic distribution, in the case where no additional component/excipient is added to the stabilized active substance.
However, the concerned manufacturers benefiting from a status of pharmaceutical establishment on all the production line must be able to keep this status, even if they conform to the remote reporting of authorization for activity of active substance manufacturing with the ANSM.

Share article
References
1. Extraits des Bonnes Pratiques de Fabrication, Bulletin officiel N° 2011/8 bis, publié en juillet 2011 par l’ANSM [59]
2. EudraLex – Volume 4 Good manufacturing practice (GMP) Guidelines: Part II – Basic Requirements for Active Substances used as Starting Materials; Part I – Basic Requirements for Medicinal Products;
3. Annex 1 Manufacture of Sterile Medicinal Products
4. Annex 2 Manufacture of Biological active substances and Medicinal Products for Human Use(171 KB)
Definitions
Active Substance : Any substance or mixture of substances intended to be used in the manufacture of a medicinal product to become active ingredients of the medicinal product;
Raw material: Any substance, active or inactive, of a defined quality used in the manufacture of active pharmaceutical ingredients, bulk materials, intermediate or finished product, whether or not it is present in the product.
End product: A product which has undergone all stages of manufacturing and packaging operations and which is in its final container.
Equivalent : DP (Drug Product)
Intermediate product: A partly manufactured material or product, which undergoes further manufacturing steps before it becomes a finished material, bulk or finished product.
Bulk product: Any pharmaceutical form (liquid, powder, suspension) that is either to be filled into another or its final container at the next process step, or is already filled into its final container to be labelled and packaged at the next process step.
Equivalent : Bulk
Glossary
BPF / GMP : Bonnes Pratiques de Fabrication / Good Manufacturing Practices
DS / API : Drug Substance / Active Pharmaceutical Ingredient ou Substance active
DP/PF : Drug Product / produit fini
AMM : Autorisation de Mise sur le Marché
ANSM : Agence Nationale de Sécurité du Médicament et des produits de santé
QP / PR : Qualified Person / Pharmacien Responsable
IMP : Investigational Medicinal Product
