


Sommaire
- Gene therapy manufacturing comes of age: Commercial-scale manufacturing is imminent. Are gene therapy innovators ready?
- Overcoming obstacles in downstream bioprocessing of AAV based gene therapy products
- Overcoming challenges in the development of lentiviral vector manufacturing platforms
- Are modern scalable bioreactors the Cell Culture Strategy needed for Gene & Cell Therapy success?
- The magnetic power of nanoparticles magnetic cell sorting decontamination of-environments and reusable nanocatalysts
- Development of a new preventive approach to reduce microbial infections with metal oxide nanoparticles
- Maximizing Sterility Assurance: Sterile Hold Time Testing for Sterilized Items Used in Parenteral Drug Manufacturing
Overcoming Challenges in the development of lentiviral vector Manufacturing Platforms
Gene therapy has become increasingly promising in recent years, with the use of viral vectors to deliver genetically modified cells that can correct genetic defects or treat diseases. Lentiviral vectors in particular received significant attention in the gene therapy space thanks to their ability to stably integrate into the genome of the host cell and allow for longterm gene expression. They have shown great promise in gene therapy, with several clinical trials demonstrating their efficacy in treating diseases, and their demand is constantly rising to support advanced clinical phases and commercial applications.

1. Lentiviral vectors: a safer and more versatile option for gene therapy
Over the course of the years viral vector-based gene therapy shifted from the use of γ-retroviral vectors derived from murine leukemia viruses (MLV) to lentiviral vectors (LV) mainly derived from HIV-1. While both retroviral vectors are able to stably integrate the transgene of interest, lentiviral vectors have the ability to transduce both dividing and nondividing cells and offer a safer genome integration profile, reducing the risks of insertional mutagenesis. At the same time, LV vectors can generally deliver higher infectious titres. The most common approach for the manufacturing of the LV vectors is the four plasmids system, each of which plays a specific role in the production of the vector. This system is widely used because it allows for precise control over the viral genome and ensures that the viral particles are replication-defective, meaning they cannot reproduce and cause disease in the target cells. The first plasmid contains the lentiviral genome, which includes the gene of interest that will be delivered to the target cells. The second plasmid contains the genes encoding for the structural proteins of the viral particle, which include the capsid, envelope, and accessory proteins. The third plasmid contains the packaging genes, which encode for the enzymes necessary for packaging the viral genome into the viral particle. The fourth plasmid contains the Rev gene, which is necessary for the transport of the viral RNA from the nucleus to the cytoplasm. The four plasmids are transfected into a producer cell line, where the lentiviral genome, structural proteins, packaging genes, and Rev gene are all expressed. This leads to the production of lentiviral particles, which can then be harvested and purified for use in gene therapy applications.
2. Current Manufacturing Platforms and their limitations. Adherent systems – The CellFactory model
One of the key considerations for lentiviral vector manufacturing is the choice of cell type and culture system. Adherent cell culture systems (HEK293T or HEK293 cells) are the most commonly used for lentiviral vector production. They require solid adhesion surfaces on which cells can grow up to optimal confluence, when they are transfected to prompt LV vector production. From small scale productions in Petri dishes, this system can be scaled up on larger supports, such as T-flasks or multitray systems like CellFactories (10-trays, 40-trays). By increasing the number of CellFactories processed in parallel (scale-out approach) this setup can lead to LV production processes that result in volumes of 20 -50L of bulk supernatant. This approach is widely known and proved effective in producing high-titre LV vector bulks.The harvested supernatant containing the LV vectors is then processed into the downstream phase, with the aim to concentrate the LV vector to high-titres and to get rid as much as possible of impurities to guarantee the safety of the product and maximize the transducing capability of the LV vectors. To this aim the harvested bulk is processed through multiple steps that include clarification, chromatographic steps for separation of the LV vector from impurities, concentration with tangential flow filtration (TFF), diafiltration or size exclusion chromatography for final formulation and a final sterilizing filtration. This multi-step purification/concentration approach has a significant cost in terms of LV recovery, with fractions of LV vectors lost at each step. Purification steps aimed at separating the LV from impurities most of the time result in non-negligible losses. Ion Exchange Chromatography is the most widely used approach, leveraging the net negative charge of the LV to separate it from BSA, residual host cell proteins, DNA and other impurities. Negatively charged molecules however tend to be bound by the resin and then eluted together with the LV vector, requiring a fine tuning of elution conditions to obtain the desired balance between purity and LV vector recovery. Moreover, the sensitivity of LV vectors to moderately high salt concentrations must be taken into account when defining elution strategies, to preserve infectivity.Resuspension of the purified LVs in the desired formulation medium can be carried out both via chromatographic approach – such as Size Exclusion Chromatography (SEC) – or via tangential flow filtration. The chromatographic approach with SEC can however be applied up to a certain scale, as the processing of larger volumes would require the use of increasingly taller columns, with the related resin packing issues and space requirements. The TFF approach consists in a diafiltration step that can often be coupled to a previous concentration phase. While LV vectors are sensitive to shear forces and could be damaged by harsh conditions, it is possible to tune the concentration and diafiltration parameters to ensure excellent recoveries while maintaining high infectivity.Finally, the sterilizing filtration can be considered the most critical step during the DSP phase. Due to their large size (approximately 100 nm), close to that of the pores of sterilizing membranes, filterability and throughput are major concerns for this step. Aggregation of LV vectors in larger complexes, or the presence of DNA fragments or proteins bound to their capsid, can increase their size, further hindering their filtration. This often results in overall poor performances of these steps, with recoveries of LV vectors across sterilizing membranes as low as 20 – 30%. Despite these challenges, processes based on the use of adherent cells grown in cell factories were developed in the last years and proved to be a reliable and effective approach for the production and purification of high quality, high potency lentiviral vectors. AGC Biologics proprietary adherent process, based on the culture of HEK293T cells on 24 x 10-trays CellFactories is able to produce 48L of clarified harvested bulks containing an average of 4.1×107 TU/mL that are purified and concentrated in the downstream phase to ~ 500 mL of high quality, high purity sterile filtered LV vectors with infectious titres of average 6.5×108 TU/mL. At the same time >99% removal of residual Host Cell Proteins, residual total DNA and BSA is achieved. In consideration of their manufacturing scale, Cell Factory based processes can support preclinical studies or early stages of therapeutic development.This approach is applied by cGMP facilities to produce clinical-grade vector preparations and are adequate for Phase I/II trials, which usually involve only a few patients. As the number of patients receiving regenerative medicines increases, both through clinical trials and approved products that have come to market, companies across the clinical development timeline are implementing strategies to deal with manufacturing and scale up. However, scaling up using cell factory and roller bottle processes can lead to significant scaling issues, where the number of supports required can result in lengthy manufacturing times, high labor costs, and increased risk due to the number of manipulations required. The scale out of these systems leads to an increase in the number of supports which determines an increase in the spaces in the laboratory and in the incubators, in the manual handling and risk of contamination from open manipulations, particularly problematic as batches are often pooled for downstream processing. Furthermore, the transfection step is multiplied for all the different supports, increasing the chance of variable vector production. In order to overcome these challenges, one alternative is to scale up the adherent cell process in a fixed-bed bioreactor. Our team at AGC Biologics has worked in the last few years to address this issue and developed a new process in bioreactor that have been successfully transferred in GMP unit, starting from our current GMP process, that foresees the transient quadri-transfection of adherent HEK293T in CFs.
3. Fixed-bed bioreactors
The fixed-bed bioreactor technology is based on the use of a compact fixed-bed filled with microfiber microcarriers with an integrated perfusion system. Primary advantages of employing a fixed-bed bioreactor technology include direct transfer of the 2D reference process which minimizes risk and saves time. Large scale fixed-bed bioreactors can reach surfaces up to 500 m2, corresponding to the growth area of approximately 800 CF10 (Cell Factory) 2D culture vessels. Moreover they allow scaling of adherent production in a controlled environment, and the highly integrated single-use equipment can be adapted to meet current good manufacturing practices requirements. The availability of representative scaled-down bioreactor models helps the development of new processes, minimizing costs and time needed to explore and optimize production parameters. In order to develop AGC 200L adherent process different parameters have been investigated such as number of cells/cm2 to be seeded, days of expansion before production, timing and number of harvest, volume of harvest, and several batches have been performed in development in order to optimized productivity and contaminants profile in a 0.5 m2 scale down model. Once the main process parameters were set, the process was scaled-up to an intermediate 1 m2 size bioreactor model, with the aim to establish a robust and reliable process for cGMP LV viral vector at 133 m2 scale, providing 200 L of bulk supernatant per batch.
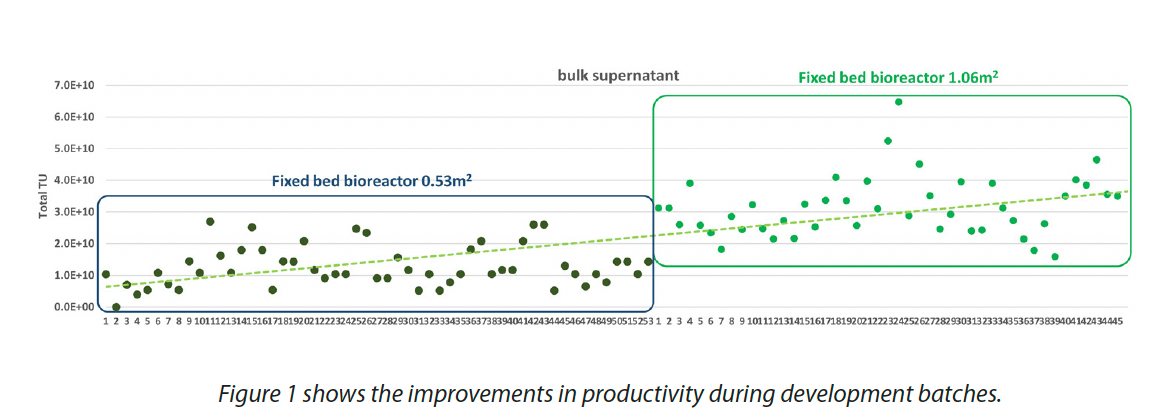 The scale up has been challenging due to the large volumes of culture medium and viral vector involved and the resulting increase in time for operations such as media change. In parallel, the increased batch size in comparison to the CellFactory setting (200L instead of 48L) poses a number of challenges to the downstream steps as well.
The scale up has been challenging due to the large volumes of culture medium and viral vector involved and the resulting increase in time for operations such as media change. In parallel, the increased batch size in comparison to the CellFactory setting (200L instead of 48L) poses a number of challenges to the downstream steps as well.
First, such larger volumes often require new, dedicated spaces and equipment such as mixers, totes, 3D-bags. The DSP equipment as well need to be capable to accommodate the increased batch size, both for TFF and chromatographic steps. For this reason, downstream processing flows applied to CellFactory-based processes often need adjustments or a complete redesign. Concentration with TFF strategy can be included as a starting step to help reduce the volumes to be further processed. While IEX chromatographic columns properly sized for full scale bioreactor processes can be easily packed or sourced from vendors offering prepacked columns, the use of Size Exclusion Chromatography is hardly applicable at this scale. Further TFF ultrafiltration steps are thus coupled with diafiltration to formulate the LV vectors in the desired medium.
Although all these challenges in scaling up the process, data obtained in the full-scale fixed-bed bioreactor system confirmed good scalability and equivalent performance of the process developed at a smaller scale as shown in Figure 2.
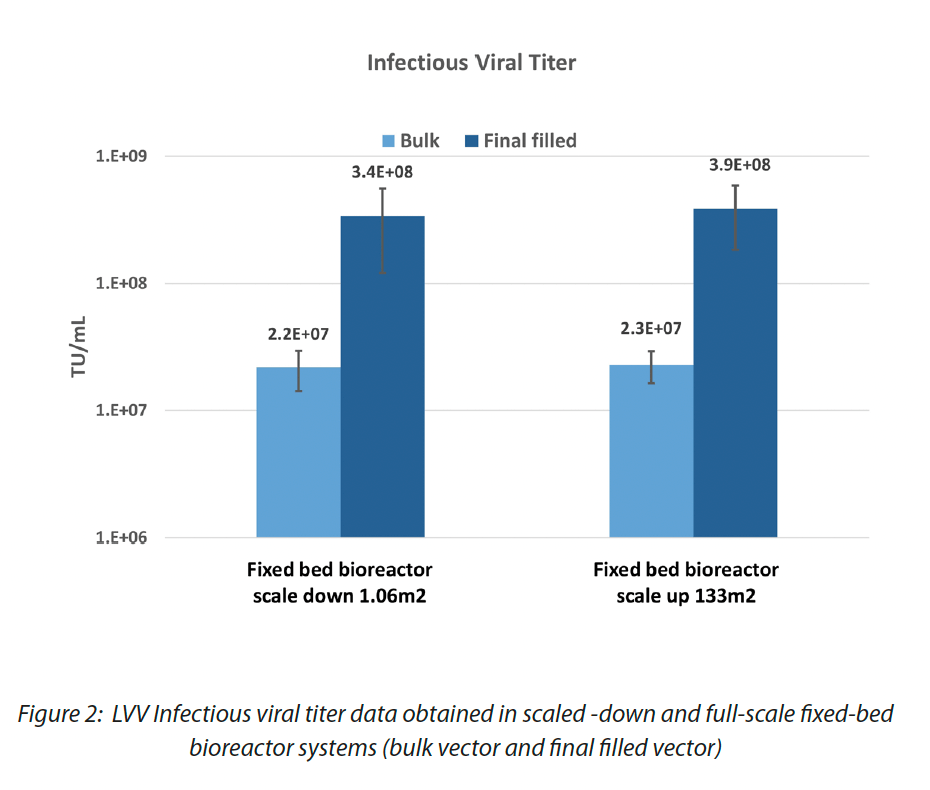
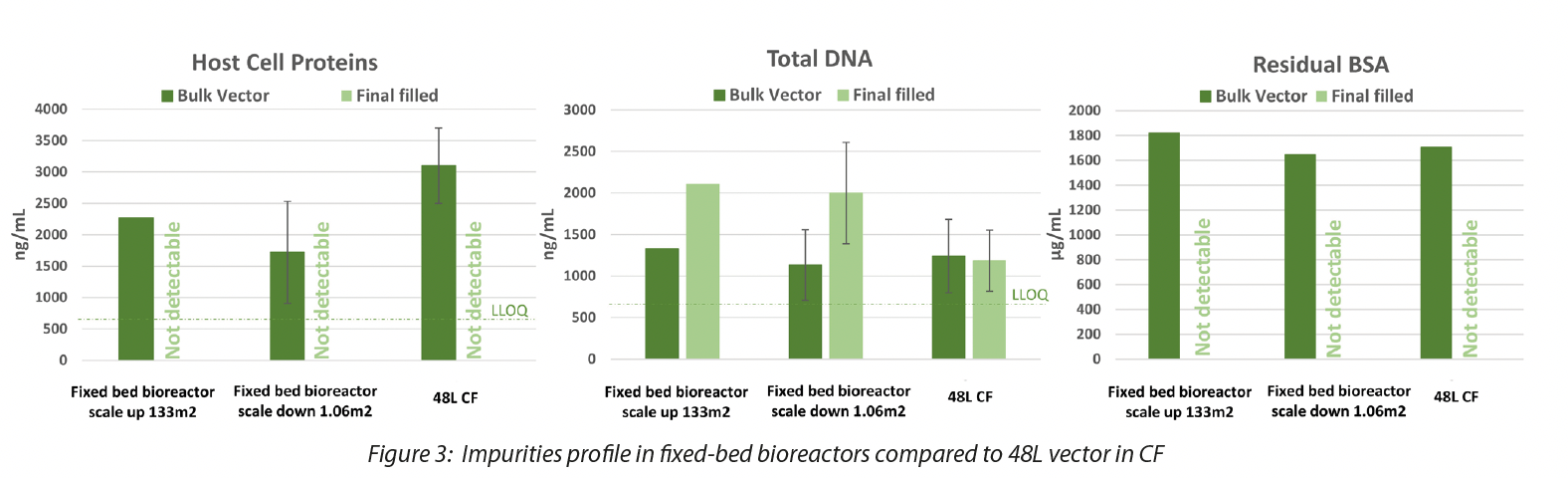
Furthermore, we demonstrated that the quality of the vector manufactured with 200L process is comparable to 48L process in terms of TU/mL and residuals as well as the transduction efficiency on target cells (CD34 hematopoietic stem cells and Lymphocytes T cells), guaranteeing an increase in the batch size (from 48L to 200L) and consequently in the total TUs of about 4 times. Data are shown in Figure 3 and Figure 4.
In AGC biologics we have also worked on a further scale up by increasing the surface of the bioreactor in order to achieve at full scale up to 1000 L of bulk vector. We are also evaluating the possibility of a further concentration of the vector in case a higher titer is needed.
 4. Suspension cell culture and Stirred-Tank (STR)
4. Suspension cell culture and Stirred-Tank (STR)
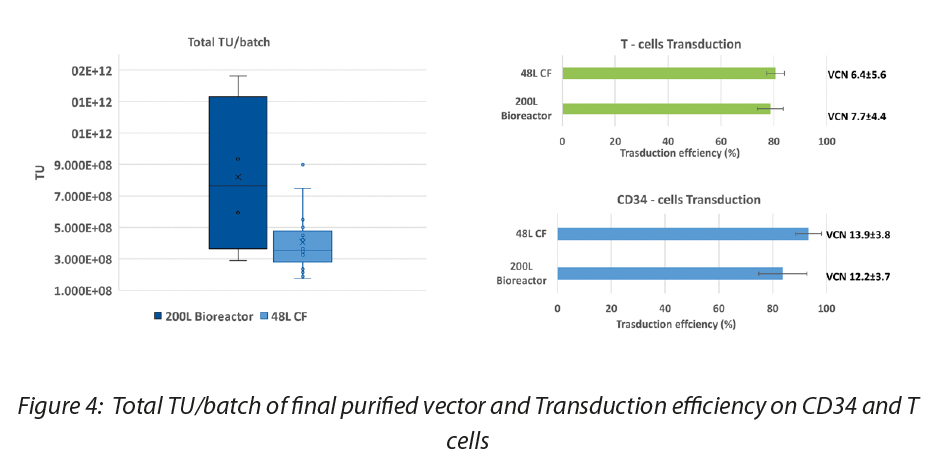
Bioreactors Although the most used platform for the production of lentiviral vectors is the one based on cells in adhesion, the system based on cells in suspension is becoming more and more interesting due to its greater simplicity in scale up. Suspension cell cultures show several advantages compared to adherent ones. Among these the greatest ease in handling the cells that do not require a tissue-culture-treated vessel to be cultured, only agitation for adequate gas exchange, can be cultivated to much higher densities in systems such as stirred tank and wave bioreactors, but above all most suspension cell lines grow in serum-free media, which reduces costs, makes the downstream purification process easier and reduces the risk of contamination with adventitious agents thus favoring the move towards a clinical grade product.
In AGC Biologics we have established a proprietary cell line by adapting adherent HEK293T cells to grow in suspension in chemically defined medium and we have developed a process for LV vector production with these cells by optimizing the culture and transfection conditions first in small scale in shaking flask, and then in STR bioreactor at 3L scale. The process was then scaled up to 50 L in pilot scale and we are now in the process of moving the process into GMP by scaling up to 200L and 1000L. Working on different process parameters, both on a small scale (for example cell density at transfection, time point for harvest, DNA/PEI ratio) and in bioreactor (stirring speed, set point of gas and pH) we managed to obtain a good yield of productivity in the bulk supernatant, even higher than with adherent system. Physical and infectious viral titers are shown in Figure 5.
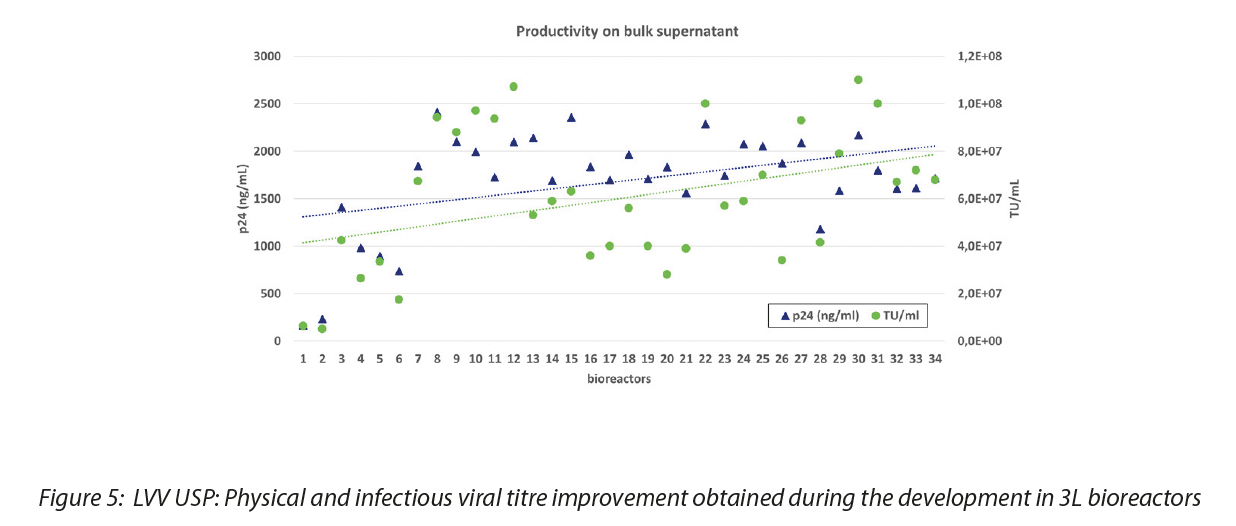
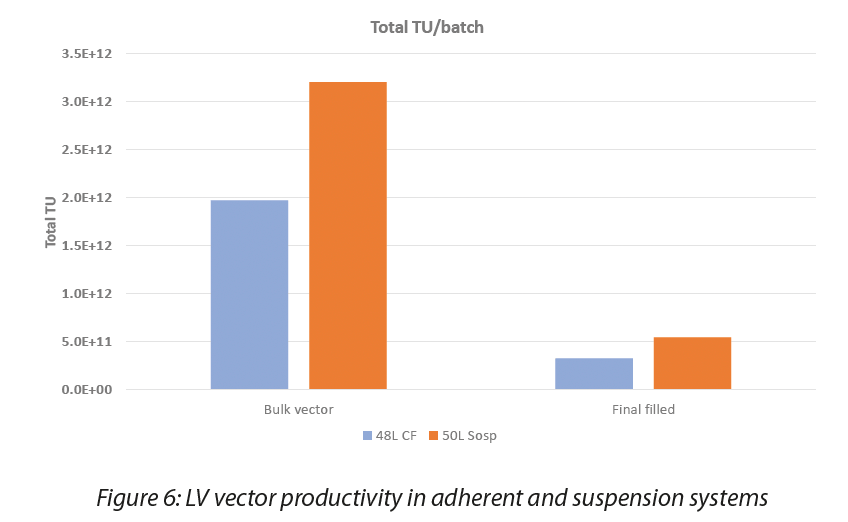
After the production and harvesting of LV supernatants, the obtained product underwent a series of purification processing steps for impurities removal and concentration. The first critical step in the downstream process is clarification of the bulk vector, to physically remove cells and cell debris from the LV-containing cell culture medium. The presence of the producer cells in suspension requires different approaches, aimed both at removing cells and cell debris and at the same time minimizing cell breakage. The use of depth filters or recently-available inline centrifugation systems can help in this regard, however clarified bulks from suspension cell systems tend to be richer in impurities such as Host Cell Proteins (HCPs) or Host Cell DNA. This poses a burden on the downstream steps that need to be tailored to the different quality of the starting material. Productivity data obtained with AGC suspension process at pilot 50L scale compared to adherent 48L process are shown in Figure 6.
5. Common challenges
Regardless of the scale and the choice of adherent vs. suspension cell systems, the establishment of LV vectors manufacturing platforms is bound to face a number of obstacles intrinsic with the specific nature and application of this kind of “advanced therapy medicinal product”.
- Representative scale-down models
The development of representative scale-down models in parallel to the definition of the full-scale process is critical for establishing robust and reliable lentiviral vector production platforms. By using these models, manufacturers can identify and address potential process issues early in the development process, thereby minimizing the risk of product failure or delay in clinical development. Additionally, at later stages, scale-down models can be used to support process validation, as they can provide evidence of the consistency and reproducibility of the manufacturing process across different scales. Constraints in the availability of equipment and consumables limit the minimum scale at which a LV vector production scale-down process can be executed, thus representing a major difficulty during development that can cause cost and project timelines to increase.
- Improving process performance
When trying to optimize LV vector productivity in the upstream phase the complex and variable nature of the starting materials, such as the host cells and culture media, must be carefully taken into consideration. Changes in the parameters or materials used in the upstream production phase can have a significant impact on the quality and consistency of the product, and in turn affect the performance of the following downstream steps. A careful balance must be reached to optimize the final product titres and recoveries. Moreover, the multi-step nature of the purification processes represents a significant challenge in terms of yield optimization, that plays on a thin balance between high yields and purity, while maintaining the stability and functionality of the lentiviral vector. New affinity chromatography resins currently under development by different manufacturers might help in this regard, possibly combining high purity with higher step LV vector recoveries. On the other hand, the substantial cost of these resins needs to be considered in the overall economy of the manufacturing process. As previously mentioned, the final sterilizing filtration represents a major burden on LV production processes yields. The introduction of larger pore size membranes as pre-filter to ease the load on the sterilizing step or the optimization of the buffer composition can be applied, even though in general this can result in only limited improvements. Moreover different lentiviral vectors might need different conditions or filter combinations resulting in longer and more expensive development work.
- Unique Analytics
Analytical testing is a crucial aspect of the production of lentiviral vectors as ATMPs, as it is necessary to ensure the safety and quality of the final product. The regulatory authorities require extensive analytical testing to be conducted throughout the manufacturing process to evaluate the identity, purity, potency, and safety of the lentiviral vectors. These tests typically include assays for determining the viral titer, transduction efficiency, residual host cell proteins and DNA contamination or any other process-related contamination. AGC LV manufacturing platforms rely on more than 100 in-house QC tests for the characterization and release of LV with orthogonal strategies. This represents a major workload and needs to be carefully considered when defining project timelines.
- Regulatory compliance
Despite there are already established guidelines and regulations in place for the use of lentiviral vectors as ATMPs, regulatory authorities continue to refine and update these guidelines as new information and experience is gained, with the aim to ensure the safety and efficacy of lentiviral vectors as ATMPs. These changes may require manufacturers to implement new or modified procedures to ensure compliance with the updated regulations. For example, changes in the required testing for safety or quality control may require additional steps in the manufacturing process or modifications to existing equipment. Additionally, changes in the regulatory environment may necessitate the development of new analytical methods or the refinement of existing ones.
- Process changes during the product life-cycle
Any change made to the manufacturing process can have a significant impact on product quality, yield, cost and acceptance by regulatory agencies. These factors can ultimately impact project timelines and time to market. Therefore, a comparability study is required to transition from one system to another, and it is crucial to consider all the potential ramifications of any changes made to the manufacturing process.
- In vivo application
Regulatory requirements for in vivo use of LV vectors are more stringent in terms of residuals levels allowed in each vector dose. This opens for the field two major challenges: from a process point of view there is the need to increase the grade of purification of lentiviral vectors, evaluating new approaches like affinity resins and membranes. In parallel, the development of analytical methods with sufficient sensitivity to detect very low concentrations of host cell proteins and host cell DNA becomes crucial.
Conclusion
Cell and gene therapies have emerged as promising treatments for previously untreatable diseases, and viral vectors are currently the preferred gene-delivery vehicle for most of these advanced therapies. As a result, safe, robust, and cost-effective manufacturing processes are necessary to meet the demands of the patient population. However, the complexity involved in developing and scaling viral vector processes to commercial manufacturing scale, as well as the lack of standardized approaches, remain as challenges that can impact therapy development timelines and productivity. Therefore, the selection of an appropriate production platform plays a key role in the successful implementation of a process that meets commercialization timelines and manufacturing costs, ultimately making these therapies accessible to patients.
Glossary
BSA Bovine Serum Albumin
CF Cell Factories
DSP Downstream Process
GMP Good Manufacturing Practices
HEK Human Embryonic Kidney
IEX Ionic Exchange Chromatography
LV Lentiviral
MLV Murine Leukemia Viruses
SEC Size Exclusion Chromatography
TFF Tangential Flow Filtration
USP Upstream Process
References
• Large-Scale Production of Lentiviral Vectors: Current Perspectives and Challenges; Pharmaceutics 2020, 12, 1051
• Lentiviral Vector Bioprocessing; Viruses 2021, 13, 268.
• Production of lentiviral vectors; Molecular Therapy — Methods & Clinical Development (2016) 16017
Partager l’article


