


Summary
- Viewpoint of the ANSM’s Inspection Division (DI) concerning the ICH Q12 guideline
- ICH Q12: the basics. Review of the work of the A3P ICH Q12 GIC
- First steps towards ICH Q12: Leveraging process understanding & development data to define process Established Conditions
- ICHQ12 Implementation from an Industry Perspective with a Focus on Established Conditions
- ICH Q12 compliance and Unified Quality and Regulatory Information Management
- Burkholderia cepacia strikes again
- New EMA Sterilization guideline Guideline on the sterilisation of the medical product, active substance, excipient and primary container (EMA/CHMP/CVMP/QWP/850374/2015)
- How to store highly sensitive drugs? The benefit of functional coatings
New EMA Sterilization guideline Guideline on the sterilisation of the medical product, active substance, excipient and primary container (EMA/CHMP/CVMP/QWP/850374/2015)
This guide replaces the “Decision trees for the selection of methods” document (CPMP/ QWP/054/98)
The draft version of this text was published for consultation in April 2016. As soon as it was published, A3P created a GIC (common interest group) to comment on this draft initially, then, following the publication of the final version, to provide additional information if necessary.

Through this GIC and following several meetings, A3P was one of the 26 companies, organizations or associations to propose changes. A3P proposed almost 80 changes and more than 50% of these were incorporated, either in whole or in part. This work helped enhance the final document in order to best address the expectations of manufacturers
The final version was approved at the end of 2018 and has been applicable since 1 October 2019.
The purpose of this article is to comment on some parts of the text and to provide clarifications to help us implement it.
Although chapter 1 introduces this document, it is nonetheless necessary to clarify certain points :
• For highly sensitive products, even if this is not expressly indicated, it is highly recommended that these be discussed with the authorities at a very early stage. In addition, in the event of a new process and/or building, it is recommended that the prior agreement of these authorities be obtained.
• The term “sterile filtration” is often used rather than “sterilising filtration”. Obviously, the objective is to perform sterilizing filtration on the product AND for it to be sterile.
Chapter 2, which defines the scope of this guidance, also requires some additional information or comments:
• Unlike in some other countries, such as the USA (FDA), it could easily be interpreted that equipment sterilization data do not need to be transmitted in the quality dossier. Obviously it is nonetheless essential to have this data available.
• Although the order of recommendation of sterilization method choices, recommended by the Ph.Eur, is not defined in this chapter, it is clear that gas sterilization comes after sterilization by ionizing radiation. This is indicated in more detail in chapter 4.1
• Although this text does not take potential contaminants into consideration, the Ph. Eur 5.1.1 clearly indicates that it is necessary to demonstrate that the process used eliminates or inactivates all potential viral contaminants.
With respect to the chapters concerning sterilization (4.1.1 to 4.1.4), while this new guidance has the merit of supplementing the regulatory arsenal available on the theme, sometimes usefully with respect to the expectations of authorities, experienced professionals could question some drafting inaccuracies or deviations from the state of the art as knowledge currently stands.
1. For example, the use of the expression “steam sterilisation process”, which in the strict sense of the term limits the scope of the text to saturated steam sterilization, but it is reasonable to consider that the writers intended this expression to cover all moist heat sterilization processes; i.e. also including superheated/overpressured water sterilization, steam-air mixture or immersion sterilization. It seemed logical to us to consider the text as being applicable to all moist heat processes.
2. 1976 GMPs already issued Fo ≥ 8-type formulas that could assure sterility, which is correct but in very rare and specific conditions. Since these conditions were inadequately specified, industry players found themselves confronted with serious difficulties following overly rapid interpretations or readings. Since then, such expressions have been avoided and systematically supplemented by all the conditions to be met. We would have appreciated a description of these conditions in the current text. The phrase “a steam sterilization process requires a sterilizing value of Fo ≥ 8 min to be obtained with biological indicators with a value D ≥ or ≤ 1.5 min” is difficult to understand as it stands and clearly warrants operational precautions…
The pharmaceutical industry experienced accidents and convictions in the 1980s following these unbounded expressions, without specification of the operating conditions.
It should be recalled that a duration of X minutes is a known time which elapses. To guarantee that we will arrive on time, it is also necessary to set the departure time.
In the case in point, it is necessary to know the identification and the count of the microbiological population (control of the bioburden) in order to ensure that the objective is reached (for example SAL ≤ 10-6).
For example : exposure of the product to the sterilizing agent for 8 min with a D121 = 2 min (≥ 1.5 Table 1 line C of the table below) results in a 4 log reduction in the bioburden.
Hence if the bioburden = 100 >> SAL = 10-4 not sterile in the regulatory sense of the term
• if 102 >> 10-2 ???
• if 104 >> 100 i.e. a SAL with 1 microorganism/unit
• if 106 >> 102 i.e. disinfection
According to line D of the same table, we should therefore have:
• A D121 value = Fo/(12 log reduction) = 8/12 = 0.66 min for the identified most heat-resistant strain to claim a reduction of 12 log : 106 >> 10-6 or
• A D121 value of 1 min in the following configuration 102 >>10-6.
All this without taking into account the variability of the parameter z quantifying the heat resistance. It is implied = 10 K in this text but only for moist heat and heat-resistant microorganisms clearly identified in their environment. It should be recalled that certain substances, in gel form for example, have enabled the detection of z= 17 K for Bacillus subtilis.
3. Since the cumulative sterilizing effects at temperatures below 121°C are relatively weak, except for thermolabile products, it is not necessary to comment on the calculation recommendations from 115°C or even 110°C with justification. Everyone is free to choose their own value.
4. Table 1: Cycles for steam sterilization and final heat treatment after an aseptic process, with the corresponding data required in the Quality file.
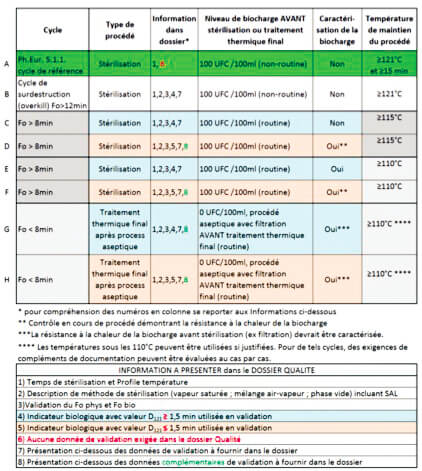
Comments
a) respective of the sterilization method (moist heat, dry heat, radiation), the whole text recommends and encourages compliance with the parameters recommended in the Ph.Eur, in return for which, although the validation data must exist, they no longer need to be presented in the quality dossier.
b) explained above with regard to the Fo value, the “15 min at 121°C or more” pairing cannot enable any industry player to assure the sterility of its product without a solid dossier and, in particular, characterization of the bioburden, which is not required in line A !!!!
It is true that in a very large number of cases it will be possible to demonstrate that the product has undergone a treatment enabling a regulatory SAL or below to be reached …
but even limiting the initial bioburden count (starting point) to 100 CFU/ml = 102, if the D value of the most heat-resistant microorganism (travel speed) in the product (for example liquid whose specific composition and/or ionic state increases this), it becomes very uncertain or even impossible to guarantee that a maximum SAL of 10-6 is obtained
For example: Fo/D= log reduction number = 15 min / (2 min/log) = 7.5 log i.e. 102 >> SAL 10-5.5 > 10-6.
c) This raises questions about the need to create 2 families of 4 lines C, D and E, F differentiated only by a calculation starting point of Fo at 115°C or 110°C, knowing that each time, in parallel, additional data is requested if less than 1.5 min. Once again the logic is strange because the lower the D value and thus the less heat-resistant the microorganism is, the higher the log reduction number will be for the same duration (F> 8) and the better sterility assurance will be.
d) Lines G and H, the rationale is critical for treatment not accumulating an Fo of 8 but fortunately characterization (implying identification) is required.
However, it can be noted : Fo/Log number = D. If Fo = 8, then for a 6 log reduction resulting in an SAL = 10-6 starting from a bioburden of 0 CFU/100 ml, D must not exceed 8/6 = 1.33, well below 1.5 (line H). If D= 1.4 (sometimes Bacillus atrophaeus in some pharma liquids), then 8/1.4 = 5.7 i.e. SAL = 10-5.7 > 10-6.
However, it is clearly specified that lines G and H describe the final heat treatment parameters after aseptic filling and not those for sterilization.
The spirit is to reinforce sterility assurance after the filling operation where possible. Whatever the number of log reductions acquired by additional complementary treatment, this will reduce the potential undetectable risk of non-sterility.
It will be necessary to validate with the authorities the cases for application of these additional treatments or if the trend is moving towards a systematic generalization.
5. As regards terminology, the meaning of expressions such as “SAL demonstrated in validation” will be understood, even if, strictly speaking, it will be remembered that a SAL value results from a mathematical extrapolation which cannot be demonstrated by physical or biological validation. The manufacturer must therefore carry out a microbiological qualification demonstrating the logarithmic reduction in the number of spores exposed to the sterilizing agent.
6. The sterilizing value Fh (Tref = 170°C; z=20K) in dry heat thermal processes has not been taken into account although the one for moist heat processes is very frequently used.
Is this due to reluctance or a fear of generating a triple safety margin!!!
7. Gas sterilization: no significant change.
Still no recognition of parametric release or elimination of routine biological indicators despite rigorous Performance Qualification. The sacrosanct “sterility test” remains despite its lack of representativeness and especially the false certainties that it can generate.
In the paragraphs relating to gas sterilization, known as surface sterilization, essential “sufficient penetration” is sometimes mentioned, which should be interpreted as gas penetration under temperature and humidity conditions inside the containers of the load, with the gas only penetrating surfaces typically permeable to air and only penetrating the surface layers of product surfaces in negligible quantities.
Pressure, although a critical parameter of the cycle, is not a process parameter because it does not affect the quality of the sterilization.
The process parameters are the gas concentration for a given time in the presence of the Temperature and Humidity variables acting as catalysts.
From chapter 4.1 defining requirements, it clearly emerges that product formulations are expected that do not leave room for any microbiological contaminants, which would not be kept to a minimum, with it not being necessary to justify the limits by filter retention capacities.
Chapter 4.1.5 specific to filtration sterilization reiterates in table form the data to be submitted in the quality dossier. This information and the parameters related to the filtration process are usually part of any good filtration validation dossier, in particular aligned with the PDA Technical Report No. 26 “Sterilizing Filtration of Liquids”, which is the reference in this field. New: the presence of non-sterilizing grade filters in the process needs to be documented in this section.
Some data, such as the evaluation or quantification of potential phenomena of adsorption or of substances extracted and released by materials in contact with the products (extractable and leachable substances) are, depending on the process configurations, to be considered over the entire product process, with filtration constituting only a sub-part.
The main novelty, or confirmation of updated expectations, relates to the verification of the integrity of the filters by tests carried out online immediately after their use. This is a strong recommendation (“should”) – i.e.: unless there is a “specifically justified and validated” reason for not doing so !”.
A reminder of the expectations in terms of controlling microbiological contamination before any filtration is detailed in a good part of this chapter, stressing the importance of the time spent on preparations/formulations before filtration, then the storage time and/or filling time to be justified.
It is recalled that in most situations a limit of 10 CFU/100 ml (with the TAMC method – Total Aerobic Microbial Count) for the bioburden before sterilizing filtration is adequate and that if a pre-filter is added (as an additional precaution) then this same limit is also applicable to a sample taken before this pre-filtration step.
From the start of chapter 4.1.6 defining aseptic processing requirements, the terms “technology to process sterile components…” and examples of usages targeting isolators and RABS (Restricted Access Barrier Systems) confirm the trend towards the ultimate disappearance of aseptic zones with “conventional” filling lines. Data supporting control of holding time and filling time, reduced to a minimum, may be required (particularly data collected during aseptic process simulation tests). In addition, in the event of durations exceeding 24 hours, these durations should be justified and supported by a risk assessment.
In Blow-Fill-Seal technology, a summary of the sterilization validation data should be provided with, in particular, fixed and justified data and limits for the bioburden of the material in line with the results of biological indicators and their resistance, in order to demonstrate, with a safety margin, that a sufficient sterility assurance level has been obtained (SAL ≤10-6) for the surface of the container.
In view of the impossibility of applying terminal sterilization to Advanced Therapy Medicinal Products (ATMP), manufacturers are referred to specific GMPs for their aseptic processing. (Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products).
Section 4.2 relating to active substances, excipients and containers clearly indicates the directives to be followed depending on the products manufactured. We can see that it specifies that the sterilization and aseptic processing of active substances are not covered by GMPs. However, their sterilization is a critical step in order to obtain the sterility assurance level of the finished product. Hence these processes must meet the same requirements as the finished products, including GMP annex 1 for products for human use.
Concerning the sterilization of sterile containers, this guideline refers us to the various ISO standards, depending on the sterilization methods chosen: sterilization by moist or dry heat, radiation and gas. In this context, the competent authorities reserve the right to conduct inspections at sites performing these sterilization activities.
It is specified that, among other data, the quality dossier for sterile containers must indicate the sterilization method and cycle. We understand here that detailed sterilization cycle parameters are not required.
Conclusion
Although this text is not perfect, it has the merit of reiterating the basic rules for the various forms of sterilization in the pharmaceutical field and of defining the minimum requirements. As described above, it contains a certain number of inaccuracies that we have attempted to comment on and/or explain through this article. We do not claim to have been exhaustive; however we believe that these in-depth points should help the pharmaceutical industry to apply and comply with this text.
Share article

