


Sommaire
- Point de vue de la direction de l’inspection (DI) de l’ANSM sur le document ICH Q12
- ICH Q12 : les fondamentaux. Retours des travaux du GIC A3P ICH Q12
- First steps towards ICH Q12: Leveraging process understanding & development data to define process Established Conditions
- ICHQ12 Implementation from an Industry Perspective with a Focus on Established Conditions
- ICH Q12 compliance and Unified Quality and Regulatory Information Management
- Burkholderia cepacia a encore frappé
- Nouveau guideline Stérilisation de l’EMA
- How to store highly sensitive drugs? The benefit of functional coatings
Nouveau guideline Stérilisation de l’EMA Guideline on the sterilisation of the medical product, active substance, excipient and primary container (EMA/CHMP/CVMP/QWP/850374/2015)
Ce guide vient en remplacement du document “Decision trees for the selection methods” (CPMP/QWP/054/98)
Le draft de ce texte est paru pour consultation en avril 2016. Dès sa parution A3P a créé un GIC (groupement d’intérêt commun) afin dans un premier temps de commenter ce draft puis, après la parution de la version définitive, d’apporter des informations complémentaires si nécessaire.

A travers ce GIC, et suite à plusieurs réunions, A3P a été parmi les 26 entreprises, organismes ou associations à proposer des modifications. A3P a proposé près de 80 modifications et plus de 50 % ont été intégrées totalement ou partiellement. Ce travail a permis de faire évoluer ce document afin qu’il réponde au mieux aux attentes des fabricants.
La version définitive a été approuvée fin 2018 et est applicable depuis le 1er Octobre de cette année.
Cet article a pour but de commenter certaines parties du texte, d’apporter des précisions afin de nous aider à le mettre en œuvre.
Même si le chapitre 1 introduit ce document, il est néanmoins nécessaire de préciser certains points :
• Pour les produits hautement sensibles, et même si ce n’est pas expressément écrit, il est vivement conseillé de discuter avec les autorités très en amont. De plus, en cas de nouveau process et/ou bâtiment, il est recommandé d’avoir un accord préalable de ces autorités.
• Le terme ” sterile filtration ” est souvent employé à défaut de ” sterilising filtration “. Bien sûr, le but est d’opérer une filtration stérilisante sur le produit ET qu’il soit stérile.
Le chapitre 2 qui définit le scope de ce texte nécessite lui aussi quelques compléments d’informations ou remarques :
• Contrairement à certains autres pays tels que les USA (FDA), il est facilement interprétable que les données de stérilisation du matériel ne sont pas à transmettre dans le dossier qualité. Bien sûr, il est néanmoins indispensable d’avoir ces datas disponibles.
• Même si l’ordre de recommandations dans les choix des méthodes de stérilisation, préconisé par la Ph.Eur n’est pas défini dans ce paragraphe, il est clair que la stérilisation à gaz vient après la stérilisation par ionisation. Ceci est plus détaillé dans le paragraphe 4.1
• Même si ce texte ne prend pas en considération les contaminants potentiels, la Ph. Eur 5.1.1 décrit clairement qu’il est nécessaire de démontrer que le procédé utilisé permet d’éliminer ou d’inactiver tous les contaminants viraux potentiels.
Au regard des paragraphes relatifs à la stérilisation (4.1.1 à 4.1.4), si ce nouveau texte a le mérite de compléter l’arsenal réglementaire sur le thème, parfois utilement vis à vis des ” Attendus par les autorités “, les professionnels expérimentés pourraient s’interroger sur quelques imprécisions de rédaction ou écarts par rapport aux règles de l’art en l’état actuel des connaissances.
1. Par exemple l’emploi de l’expression ” steam sterilisation process ” qui stricto sensu limite le champ du texte à la stérilisation à vapeur saturante mais il est raisonnable de penser que dans l’esprit des rédacteurs, cette expression devrait couvrir toutes les applications des procédés stérilisants à la chaleur humide (moist heat sterilisation processes) ; c’est-à-dire incluant aussi la stérilisation par l’eau surchauffée/surpressée, celle en mélange d’air-vapeur ou par immersion. Il nous a semblé logique de considérer le texte comme applicable pour l’ensemble des procédés à la chaleur humide.
2. Déjà les GMP de 1976 avaient émis des formules de type Fo ≥ 8 qui pouvait assurer la stérilité, ce qui est exact mais dans des conditions très rares et spécifiques. Celles-ci n’ayant été qu’insuffisamment précisées, des lectures ou interprétations trop rapides ont conduit des industriels devant de graves difficultés. Depuis ces expressions sont combattues et systématiquement enrichies de l’ensemble des conditions à respecter. On aurait apprécié la description de ces conditions dans le texte actuel. La phrase ” un procédé de stérilisation à la vapeur exige d’atteindre une valeur stérilisatrice Fo ≥ 8 min ” avec des indicateurs biologiques ayant une valeur D ≥ ou ≤ à 1,5 min ” est difficile à comprendre en soi et mérite bien des précautions opérationnelles…
L’industrie pharmaceutique a connu des accidents et condamnations dans les années 80 suite à ces expressions non bornées, sans précisions des conditions opératoires.
Rappelons qu’une durée de X minutes est un temps connu qui s’écoule. Pour garantir que nous allons arriver à l’heure, il est aussi nécessaire de définir l’heure de départ.
Dans le cas qui nous préoccupe, il est nécessaire de connaitre l’identification et le dénombrement de la population microbiologique (maîtrise de la biocharge / bioburden) afin d’assurer que l’objectif soit atteint (par exemple SAL ≤ 10-6).
Pour exemple : une exposition du produit à l’agent stérilisant 8 min avec un D121 = 2 min (≥ 1,5 Table 1 ligne C du tableau ci-après) entraîne une réduction de 4 log de la biocharge.
Donc si biocharge = 100 >> SAL = 10-4 non stérile au sens réglementaire
• si 102 >> 10-2 ???
• si 104 >> 100 soit un SAL avec 1 micro-organisme /unité
• si 106 >> 102 soit une désinfection
Il faudrait donc selon la ligne D du même tableau avoir :
• Une valeur D121 = Fo/(12 réduction de log) =8/12=0,66 min pour la souche identifiée la plus thermorésistante pour prétendre à une réduction de 12 log : 106 >> 10-6 ou
• Une valeur D121 de 1 min dans la configuration suivante 102 >>10-6.
Le tout en faisant abstraction de la variabilité du paramètre z quantifiant la résistance à la chaleur. Il est sous-entendu =10 K dans ce texte mais seulement pour la chaleur humide et des microorganismes thermorésistants clairement identifiés dans leur milieu. Rappelons que certaines molécules, en gel par exemple, ont permis la mise en évidence de z= 17 K pour un Bacillus subtilis.
3. Les effets stérilisants cumulés aux températures inférieures à 121°C étant relativement faibles, sauf pour les produits thermolabiles, il n’est pas nécessaire de commenter les recommandations de calculs à partir de 115°C voire 110°C avec justification. Chacun est libre de choisir sa valeur.
4. Tableau 1 : Cycles pour la stérilisation à vapeur et le traitement thermique final après procédé aseptique, avec les données correspondantes exigées dans le dossier Qualité.
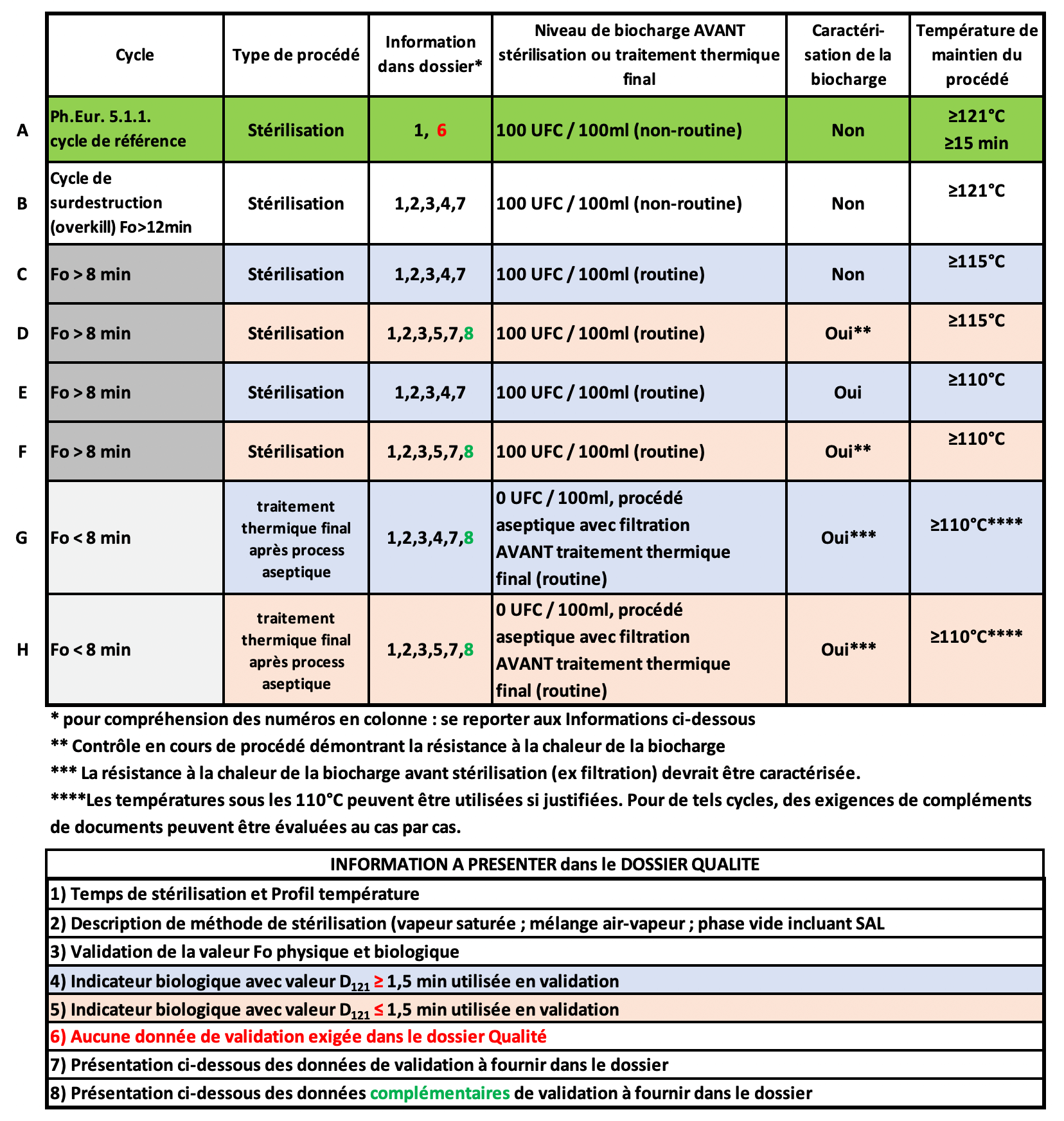
Commentaires
a) L’ensemble du texte recommande et encourage, quelle que soit la méthode de stérilisation (chaleur humide, sèche, rayonnements) le respect des paramètres préconisés dans la Ph.Eur , en contrepartie de quoi, bien que les données de validation doivent exister, elles ne sont plus à présenter dans le dossier qualité.
b) Comme explicité précédemment au regard de la valeur Fo, le couple ” 15 min à 121°C ou plus ” ne peut permettre à aucun industriel d’assurer la stérilité de son produit sauf avec un dossier solide et notamment une caractérisation de la biocharge, ce qui n’est pas exigé en ligne A !!
Certes dans un très grand nombre de cas, on pourra démontrer que le produit a subi un traitement permettant d’atteindre un SAL réglementaire ou inférieur, …
mais même en limitant le dénombrement de la biocharge initiale (point de départ) à 100 UFC/ml =102, si la valeur D du microorganisme (vitesse de parcours) le plus thermorésistant dans le produit (par exemple liquide dont la composition spécifique et /ou ionique accroît celle-là), il devient très incertain voire impossible d’assurer l’obtention d’un SAL max de 10-6.
Ex : Fo/D= nbre de réduction de log = 15min / (2 min/log) = 7,5 log soit 102 >> SAL 10-5,5 > 10-6.
c) On peut s’interroger sur la nécessité de créer 2 familles de 4 lignes C, D et E, F différenciées uniquement par un départ de calcul de Fo à 115°C ou 110°C , sachant qu’à chaque fois, parallèlement, il est demandé des données complémentaires si la valeur D de l’indicateur biologique est inférieure à 1,5 min. Là encore la logique est étrange car plus la valeur D est faible, donc le germe moins thermorésistant, plus le nombre de réduction de log sera grand pour une même durée (F>8) et meilleure sera l’assurance de stérilité.
d) Lignes G et H, le raisonnement est critique pour un traitement ne cumulant pas un Fo de 8 mais heureusement la caractérisation (sous-entendu identification) est exigée.
Toutefois on notera : Fo/ Nbre Log = D. Si Fo=8, alors pour 6 log de réduction conduisant à un SAL = 10-6 en partant d’une biocharge 0 UFC/100ml, D ne doit pas dépasser 8/6 = 1,33 bien inférieur à 1,5 (ligne H). Si D= 1,4 (parfois Bacillus atrophaeus dans certaines substances pharma liquide) alors 8/1,4 = 5,7 soit SAL = 10-5,7 > 10-6.
Il est toutefois bien précisé que les lignes G et H décrivent les paramètres de traitement thermique finaux post remplissage aseptique et non ceux d’une stérilisation.
L’esprit en est de renforcer l’assurance de stérilité après l’opération de remplissage lorsque c’est possible. Quel que soit le nombre de réduction de log acquis par un traitement complémentaire additionnel, celui-ci diminuera le risque potentiel non détectable de non stérilité.
Il conviendra de valider avec les autorités les cas d’application de ces traitements complémentaires ou si la tendance s’oriente vers une généralisation systématique.
5. En terminologie, on comprendra le sens des expressions de type ” SAL démontré lors de la validation ” même si stricto sensu on se souviendra qu’une valeur de SAL résulte d’une extrapolation mathématique non démontrable par la validation physique ou biologique. L’industriel devra donc réaliser une qualification microbiologique démontrant la réduction logarithmique du nombre de spores exposés à l’agent stérilisant.
6. La valeur stérilisatrice Fh (Tref = 170°C ; z=20K) en procédé thermique à chaleur sèche n’a pas été pris en considération bien que celle en chaleur humide soit très fréquemment utilisée.
Réticence ou crainte engendrant une marge de sécurité triple !!!
7. Stérilisation par les gaz : aucune évolution significative.
Toujours pas de reconnaissance de la libération paramétrique ni suppression des indicateurs biologiques en routine malgré une Qualification de Performance rigoureuse. Le sacro-saint ” essai de stérilité ” demeure malgré son manque de représentativité et surtout les fausses certitudes qu’il peut engendrer.
Dans les paragraphes relatifs à la stérilisation gaz, dite stérilisation de surface, il est parfois mentionné ” une pénétration suffisante essentielle ” qu’il convient d’interpréter comme pénétration des gaz en conditions de température et humidité à l’intérieur des cartons de la charge d’articles ; le gaz ne pénétrant que les surfaces typiquement perméables à l’air et qu’en quantité négligeable les couches superficielles des surfaces des produits.
La pression, bien que paramètre critique du cycle, n’est pas un paramètre de process car cela n’impacte pas sur la qualité de la stérilisation.
Les paramètres de procédé sont la concentration en gaz pendant un temps donné en présence des variables Température et Humidité agissant comme catalyseurs.
Dès le chapitre 4.1 définissant les requis, il apparaît clairement que l’on s’attend à des formulations de produits qui ne laissent pas la place à des contaminants microbiologiques qui ne seraient pas maintenus au minimum, les limites ne devant pas être justifiées par les capacités de rétention des filtres.
Le chapitre 4.1.5 spécifique à la stérilisation par filtration reprend, dans un tableau, les données à soumettre dans le dossier qualité. Ces informations et paramètres liés au process de filtration font habituellement partie de tout bon dossier de validation de la filtration, en particulier aligné avec le PDA Technical Report No. 26 ” Sterilizing Filtration of Liquids ” qui fait référence en la matière. Nouveauté : la présence dans le procédé des filtres de grade non stérilisant est à documenter dans cette section.
Certaines données comme l’évaluation ou la quantification des potentiels phénomènes d’adsorption ou de substances extraites et relarguées par les matériaux en contact avec les produits (extractable and leachable substances) sont, selon les configurations des process, à considérer sur l’ensemble du procédé produit, la filtration ne constituant qu’une sous-partie.
La principale nouveauté, ou confirmation des attendus actualisés, porte sur les vérifications de l’intégrité des filtres par des tests réalisés en ligne immédiatement après leur utilisation. C’est une forte recommandation (” should “) – c’est-à-dire : ” à moins d’avoir une très bonne raison dûment justifiée et validée de ne pas faire ! “.
Le rappel des attentes en matière de maîtrise de la contamination microbiologique avant toute filtration, est détaillée sur une bonne partie de ce chapitre, en insistant sur l’importance du temps consacré aux préparations/formulations avant filtration, puis du temps de stockage et/ou du temps de remplissage à justifier.
Il est rappelé que dans la plupart des situations une limite de 10 CFU/100 ml (avec méthode TAMC – Total Aerobic Microbial Count.) pour le bioburden avant filtration stérilisante est adéquate et que si un pré-filtre est ajouté (comme précaution supplémentaire) alors cette même limite est également applicable à un prélèvement réalisé avant cette étape de pré-filtration.
Dès le début du chapitre 4.1.6 définissant les requis du remplissage aseptique, les termes de ” technology to process sterile components … ” et les exemples d’utilisation ciblés sur les isolateurs et les RABS (Restricted Access Barrier Systems) confirment la tendance de la disparition à terme des zones aseptiques avec lignes de remplissages dites conventionnelles. Les données supportant la maîtrise des temps de stockage et de remplissage réduits au minimum peuvent être demandées (notamment des données collectées durant les tests de simulation du process aseptique). En plus, en cas de durées supérieures à 24 heures, ces durées doivent être justifiées et couvertes par une analyse des risques.
La technologie du formage-remplissage-scellage (Blow-Fill-Seal) suppose de fournir un résumé des données de la validation de la stérilisation avec en particulier les données et limites fixées et justifiées de la charge microbiologique du matériau en accord avec les résultats des bio-indicateurs et leur résistance pour démontrer, avec une marge de sécurité, l’obtention d’un niveau de stérilité suffisant (SAL ≤ 10-6) de la surface des contenants.
L’impossibilité d’appliquer une stérilisation terminale aux Médicaments de Thérapie Innovante (ATMP – Advanced Therapy Medicinal Products) est renvoyée vers les GMP spécifiques pour leur remplissage aseptique. (Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products).
Le paragraphe 4.2 relatif aux substances actives, excipients et containers nous indique clairement les directives à suivre en fonction des produits fabriqués. Nous pouvons y voir préciser que la stérilisation et les procédés aseptiques des substances actives ne sont pas couverts par les GMP, cependant, leur stérilisation est une étape critique afin d’obtenir le niveau d’assurance de stérilité du produit fini. Aussi ces process doivent répondre aux mêmes exigences que les produits finis, incluant les BPF, annexe 1 pour les produits à usage humain.
Concernant la stérilisation des contenants stériles, ce guide nous renvoie aux différentes normes ISO selon les méthodes de stérilisation choisies : stérilisation par les chaleurs humide ou sèche, les rayonnements et les gaz. Dans ce cadre, les autorités compétentes se réservent le droit de réaliser des inspections des sites réalisant ces activités de stérilisation.
Il est précisé que le dossier qualité des contenants stériles doit indiquer entre autres la méthode et le cycle de stérilisation. Nous comprendrons ici que les paramètres détaillés du cycle de stérilisation ne sont pas exigés.
Conclusion
Même si ce texte n’est pas parfait, il a le mérite de reprendre les règles de base des différentes formes de stérilisation dans le domaine pharmaceutique et de définir les minima requis.Comme décrit ci-dessus, il comporte un certain nombre d’imprécisions que nous avons tenté de commenter et/ou expliciter à travers cet article. Nous ne prétendons pas avoir été exhaustifs, néanmoins nous pensons que ces points approfondis doivent aider l’industrie pharmaceutique à appliquer et respecter ce texte.
Partager l’article

