


Summary
- How can release be accelerated via CAPA?
- Release driven and controlled by visual management
- Electronic batch record: the opportunities… and the pitfalls to be avoided
- The challenge of digitalizing production records
- AQU@Sense MB: A technological evaluation for « on-line » pharmaceutical water analysis
- Individual closed isolators for cell therapy
- Evaluation of a Quaternary Ammonium Ready To Use (RTU) Disinfectant and Hydrogen Peroxide/Peracetic Ready To Use (RTU) Combination Sanitization Regimen for Cleanroom Start-Up
How can release be accelerated via CAPA?
The goal of a pharmaceutical company is to sell drugs. This short sentence is in fact loaded with meaning and can be understood in different ways. On the one hand, production, sales, and marketing staff hear sales and economic performance, while on the other, quality and regulatory affairs staff hear drugs and compliance with regulations. Does the pharmaceutical industry have two customers?

No, the pharmaceutical industry has just once customer: the patient. As the latter is not sufficiently competent to assess quality themselves, they therefore place the efficacy and safety of what they consume blindly in the hands of the regulatory bodies that will approve (or not) the production and marketing of drugs.
It is in this dichotomy that our two categories of expert face each other every day: one setting the daily challenge of making their workshop more flexible and more productive, the other of ensuring traceability, reproducibility, and compliance to the highest standard.
Are these ideas contradictory? Not at all.
1. Are GMP and Lean incompatible?
Management of performance in the industry has been largely dominated in recent years by Lean manufacturing methods. The experience described in this article shows that these methods significantly complement Good Pharmaceutical Manufacturing Practices.
1.1 GMP, Deviations and CAPA
One of the basic frames of reference of the pharmaceutical industry is “Good Manufacturing Practices” or GMP. This is a set of rules, established by drug safety or health safety agencies, which guarantee that medicines are of high quality and ensure they are safe for users and consumers.
These rules impose a particular requirement for the monitoring of products and processes. Any divergence from expectations, any malfunction, will be declared a “deviation”. Deviations will be the subject of investigations which will lead to the setting up of corrective or preventive actions to prevent them recurring. This principle of corrective and preventive action, whose goal is to make products and processes more reliable, currently goes by the name CAPA (Corrective Action & Preventive Action, see chapters 1.4 and 1.8 of the ANSM GMP).
In addition to these principles, GMP require that deviations and the CAPA pertaining to them are traceable, ensuring that products placed on the market pass through an effective quality process. In this way, a product can be released only if no deviation occurred in the process through which it passed, or if the impact of any such deviation was evaluated and CAPA implemented.
Although GMP formally require that deviations are demonstrated, and they require investigations to be carried out to evaluate the impact on the product, and the implementation of corrective measures, they leave the manufacturer a free choice regarding the method to be adopted to do this.
1.2 Continuous improvement
The implementation of continuous improvement and more specifically of Lean Manufacturing techniques provided the opportunity to improve the efficacy of the quality assurance system. Several principles, methods and techniques were used.
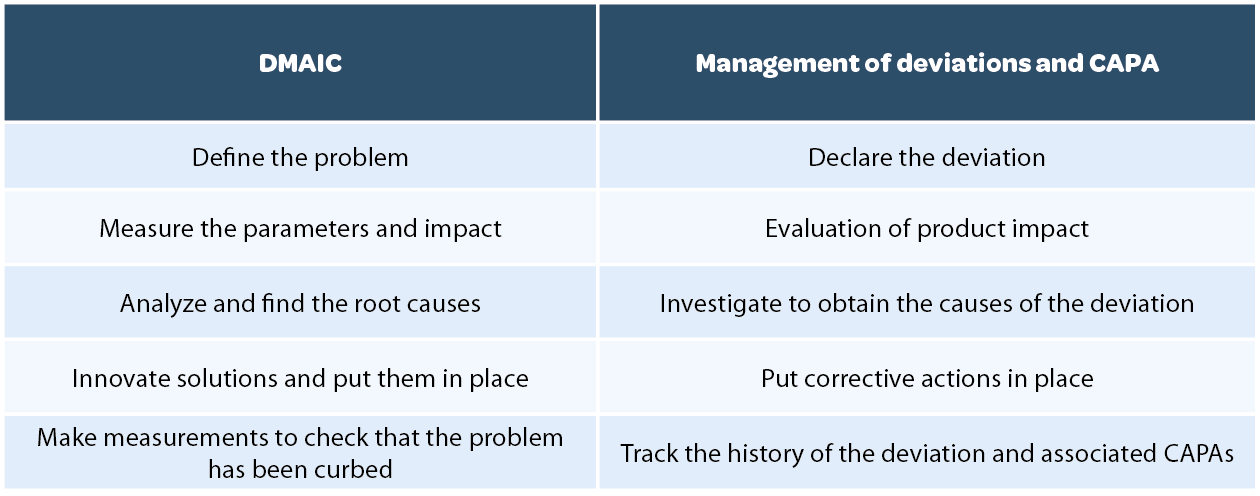
1.2.1 DMAIC
Lean manufacturing formalized the method referred to as DMAIC which consists in defining a problem, measuring parameters and impact, analyzing the root causes, implementing solutions and checking that the problem has been curbed by modifying the process in question. In the context of this article, the implementation of Lean and of the DMAIC method provided the opportunity to structure and formalize the investigative methods that generate CAPA. Indeed, if we compare the CAPA and DMAIC approaches a marked parallel is demonstrated.
1.2.2 Visual management and SIM
Visual management consists in holding very short stand-up meetings in the place of activity. These regular meetings conducted at high frequencies are also called a management ritual. The prop for these meetings is a table or a visual object that allows the quantity and nature of the problems to be resolved to be assessed in a few seconds.
The most classic tool in this field is SIM: Short Interval Management. The goal of these daily meetings is to demonstrate problems arising that day or the previous day and to define those that will be handled by the DMAIC method.
1.2.3 The process approach and breakthrough mode
In addition to the DMAIC method or using an approach that integrates this method, it may be useful to examine a process from all sides, to analyze it, understanding weaknesses and improving its efficacy. This examination will generally be carried out in accordance with a formalized method: Breakthrough Kaizen. Breakthrough Kaizen consists in bringing together all players involved with a process for a period that varies from two to five days. The players are then detached from their operational activity and dedicate themselves to the process until the expected improvement is immediately and effectively implemented.
1.2.4 Fluidity of an administrative process and the FIFO method
As with all processes, an administrative process has input information that is transformed into output information.
When a process is clogged it is tempting to manage flow with a priority system. As we will see in paragraph 4.1, changes of priority lead to malfunctions and the process becomes chaotic. Priority management ends up polluting and corrupting the normal process.
What is recommended in the continuous improvement field, is to have smooth-running processes in which the order in which records arrive is consistent with customer needs, where the first incoming records are the first to leave, following the FIFO method (First In First Out), with the priority approach remaining a very exceptional option.
1.3 Complementarities and commonalities shared by GMP and continuous improvement
In paragraph 1.6, the ANSM GMP reinforced by the ICH Q10 recommendations in part B, promote a quality system that should allow the continuous improvement of products and processes. There we find common ground with continuous improvement which is concerned as much with the product as the process which permitted its manufacture.
As we have already shown, the DMAIC method is a way of managing deviations and CAPA. But the complementarity goes further, in fact the search for root causes or investigations to look for the cause of the deviation naturally refer to the process or procedure whose malfunction generated the quality defect or the deviation. This approach by process is found equally in GMP and in the Lean manufacturing continuous improvement principles.
2. Management of product release process
2.1 Processes and principles
To improve the quality management process in a sustainable manner, it is necessary to break down the quality flows which apply to a pharmaceutical production unit and which affect the availability of the product to the customer.
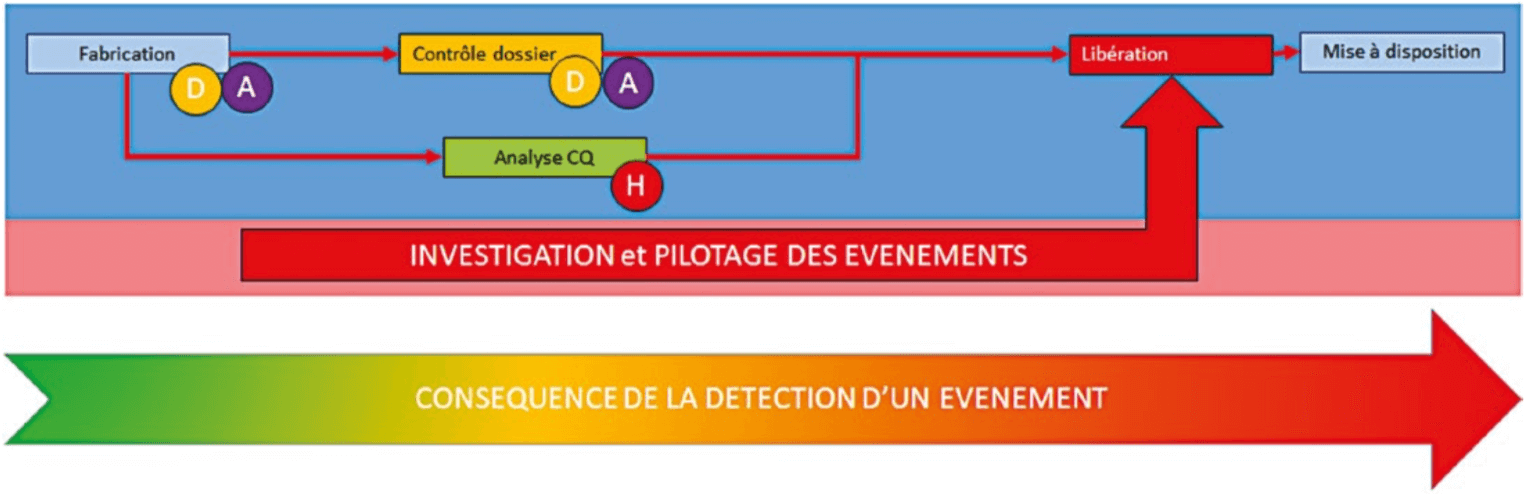
The first is the main flow (Fig.1).
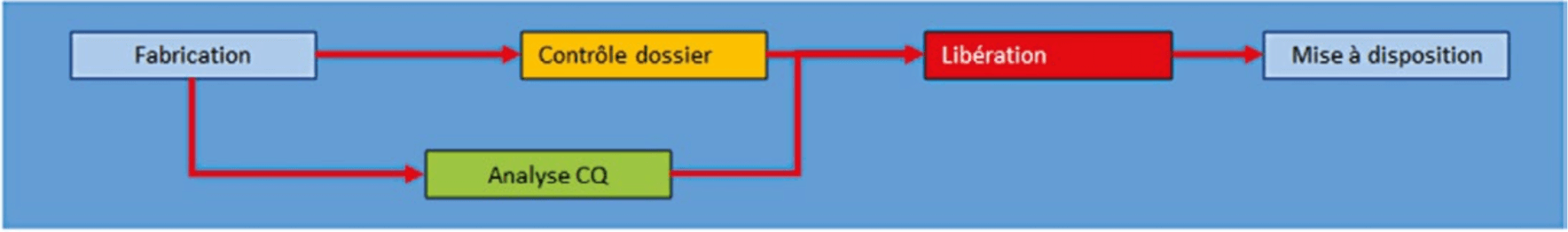
It is this that will allow product manufacture, analysis, and release. Each step adds value to product control, to allow its release. If this flow is not under control, the product release process will be affected and all staff will expend all their energy in compensating for malfunctions.
Once the batches are manufactured, the product records are reviewed. The products are analyzed and all are released to make the product available. The average durations of each of these steps must be known to be able to guarantee a reliable provisional availability date.
If all steps passed off without problem, it would be child’s play to make a production unit operate correctly and the CAPA system would not be necessary. We have to reckon with the management of malfunctions:
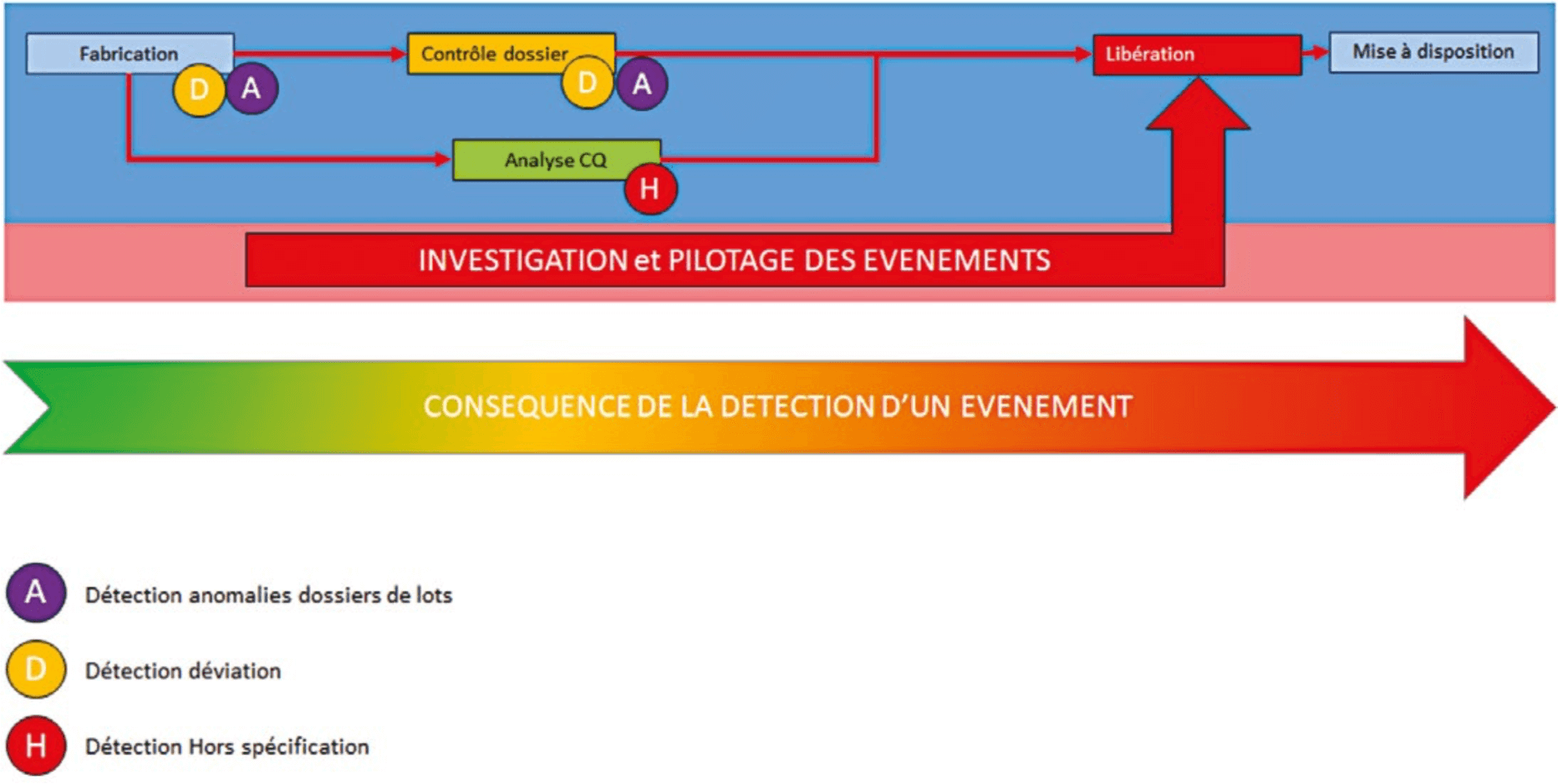
At each step, a malfunction may occur. This malfunction will have an impact on product release according to the evaluation of its impact on product quality.
The closer the occurrence of the malfunction to the planned availability date, the more it will be affected by this malfunction.
In order not to disorganize the main flow and to provide reliable availability dates, an effective management system is necessary to manage these malfunctions, to record them, carry out investigations and close them. An effective and reproducible management process will avoid disorganization of the main flow.
Good management of malfunctions is unfortunately not sufficient for production unit quality and stability to be sustainable over time. In effect, excess activity, staff absences, significantly stressful conditions, a wave of operatives leaving, requiring replacement and training, all these conditions can generate additional deviations which, added to the daily load will lead to destabilization of the main flow.
It is here that CAPAs come in:
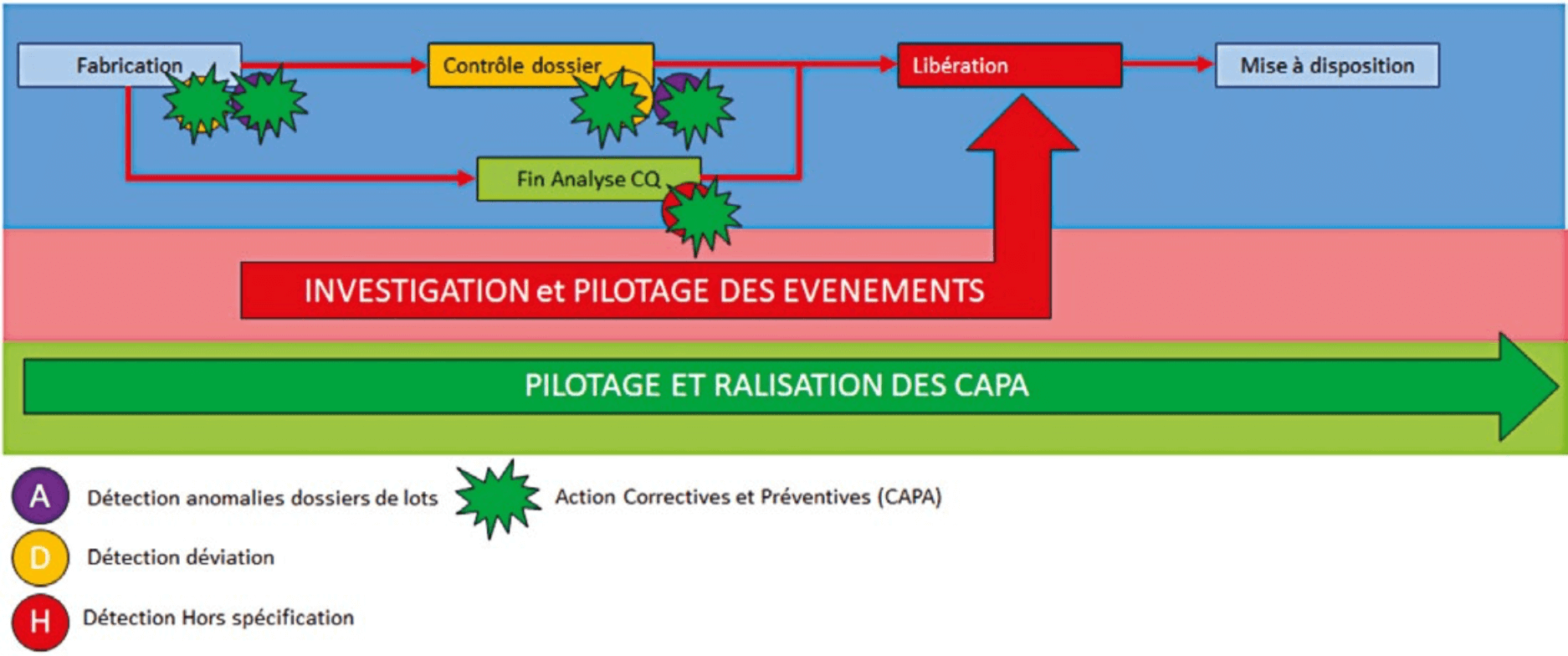
Once in place, the CAPA system will work in parallel with other systems to durably reduce the occurrence of malfunctions.
The more effective the CAPAs that are implemented, the fewer malfunctions there will be and the more time there will be to work on CAPAs and continuous improvement.
This virtuous circle is fragile, however, especially during the first months of implementation. Disruption of the main flow or of the malfunction management flow will lead to a lack of interest on the part of players in improving the flow.
Management’s quality culture is also an extremely important factor in installing the CAPA system. In fact, CAPAs do not have an immediate effect on process efficacy. CAPAs must form part of a broader management system that incorporates key indicator management (KPI: Key Performance Indicators) combined with a good knowledge of Good Manufacturing Practices (GMP). Without this overall approach, CAPA management could appear to be an administrative obligation, which it would be tempting to abandon in favor of daily management of emergencies.
The CAPA system has positive effects on production malfunctions but these effects are not exclusive to this area. All improvement activity is based on the model of the kind that involves observation, action planning, compliance with deadlines. The establishment of a true CAPA culture allows this improvement mode to be put into practice. Once this model is rooted in daily routine, it enters company culture and will benefit other areas based on the same model. Health-Safety Environment, project management and performance can be cited.
3. Starting situation, state of play
Quality assurance used to delay the release of products onto the market. Availability dates were unreliable, records awaiting release accumulated, pending the end of malfunction investigations. This excessive uncertainty regarding release dates completely destabilized the primary flow. Management of malfunctions and particularly continuous improvement increasingly fell behind schedule.
After a rapid study of the bottlenecks in department flows, it was demonstrated that the main cause lay in the fact that many records were waiting for release. Several reasons can at first glance explain this:
- A significant delay of records to be reviewed and released,
- Some records, sometimes the most eagerly awaited contain deviations currently under investigation.
By tradition release and investigation are managed and executed exclusively by quality assurance.
These tasks are described by those who carry them out as complicated, reserved for specialists, requiring great mastery, a long authorization process, with records with variable processing times which are unpredictable depending on record complexity.
If quality assurance and production were not working together to resolve problems, to prevent them returning and to anticipate new ones, the situation risked deteriorating rapidly.
4. Improvement of the process
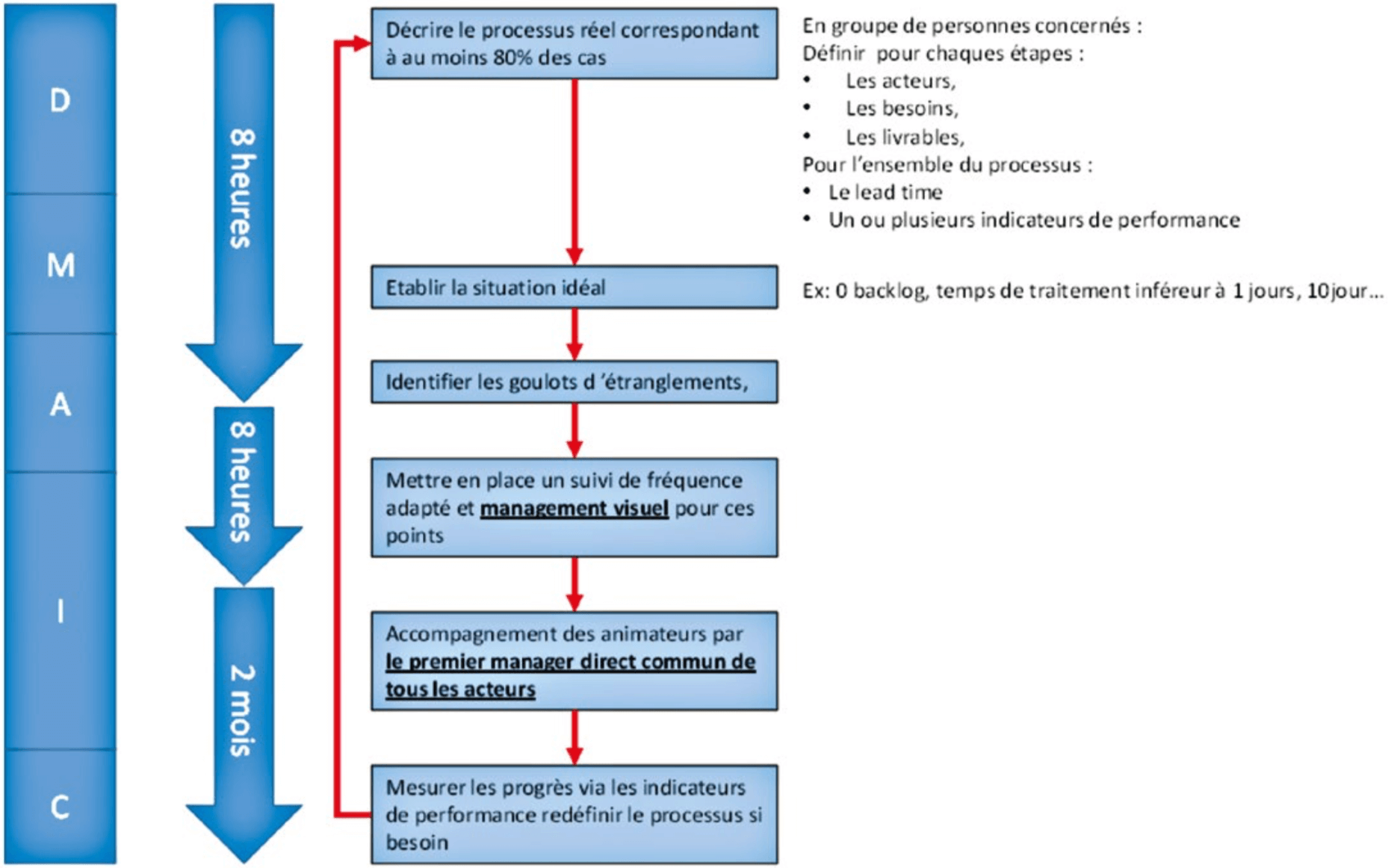
We will see how to apply the method shown above to primary and secondary flow processes in their turn and finally to processes outside the flow of a production unit to allow players to increase in competence, organization, and efficacy in order to implement an effective CAPA system.
After having described the operation of the process, the situation that is expected must be described. This is the situation that would be present if everything operated correctly in terms of time and volume. For example, no delay AND an average processing time of less than 10 days AND no more than 15 new records a day.
This information will allow evaluation of whether the capacity is appropriate given the objective.
The clarification of players, input needs, deliverables and deadlines is essential. These elements allow the establishment of an ideal situation, of performance indicators and an appropriate management system.
4.1 Batch record review activity, the first hurdle to cross
The record review process is managed exclusively by quality assurance. To overhaul its administration, it was necessary to understand fully the elements that make it up and what makes it inefficient.
The batch record is a document that supports production and which is fleshed out as and when production-related information is incorporated in it: production date and time, quantities of materials used, deviations or specific malfunctions, etc.
Insofar as this record supports production, a product is only released for the market if the batch record is complete, checked, and compliant with GMP requirements. This record is managed by staff in the quality assurance teams.
In periods of administrative overload or pressure from a customer who has run out of product, it proves counter-productive, although tempting, to put in place a priority system where the record of the most urgent product would be systematically processed as a matter of priority.
Regular prioritizing leads to malfunctions where the non-priority products of the day become priority subsequently, and one thing leading to another, the process becomes chaotic, priority management finishing by polluting and corrupting the normal process.
We have worked on this process from two angles: that of flow management and that of management of batch record anomalies.
4.1.1 Flow management
An analysis of the overall load allows us to evaluate several things:
- The average time taken to inspect each type of record,
- The ratio of the total number of records corrected per day against the number of days worked
- That the load is compatible with the number of technicians available
- A minimum quota to be completed per day to return to a stable and acceptable number of records outstanding.
If the load is compatible with the number of technicians, the goal is to have a level of responsiveness sufficient to absorb the incoming flow and therefore to put in place a “first in first out” (FIFO) system for the review of batch records.
If the load is incompatible with the number of technicians, several levers are available, such as the simplification of batch records or a streamlined review which can allow the process to be initiated while reducing inspection time and therefore increasing the number of records reviewed per day.
Working in “Breakthrough Kaizen” mode (See Paragraph 1.2.3) making batch record review the priority of QA technicians allows the priority management system to be replaced by the FIFO method in less than one month (see paragraph 1.2.4). This method makes the process more fluid by reducing the “pending” level and optimizing technician workload.
4.1.2 Management Ritual
As a medium for the organization described above we have put in place a ritual based on SIM visual management. (See paragraph 1.2.2). As we will see in the next part of this article the dashboard was fleshed out throughout our improvement initiative and began with the setting up of an input/output/remaining indicator allowing the activity to be kept under control.
4.1.3 Management of anomalies
The anomalies (“A” on the diagram) and their corrections, which are another significant item that wastes time in record reviews.
We expanded the visual management ritual described above (in paragraph 4.1.2) by incorporating management of anomalies in the dashboard, which was made visible with a daily review indicator in the presence of sector supervisors. Thus the anomalies were largely corrected in less than 24 hours.
4.1.4 Benefits obtained in batch record management
This management releases up to 50% of technician time, which was invested directly in training operatives in batch record completion, so reducing anomalies and therefore improving the document anomaly indicator and assigning training in record completion to quality assurance and not to the most experienced operative on the line (time saving in training on the line). This time was also invested in the investigation of production deviations, initially carried out by the production pharmacist.
4.2 Deviation management, at the heart of the disorganization
4.2.1 Deviation flows, state of play
A record and therefore a batch cannot be released if it is impacted by a malfunction such as deviations, for example.
Deviations heavily penalize the main flow and activity, (See paragraph 3.1).
Deviations are detected during operations and are the subject of a declaration recorded by the quality assurance teams in a digital management system: the QMS (Quality Management System).
In order to release a product, each impact of the deviation on the product must be analyzed and a decision must be made on whether it should be the subject of a CAPA.
The first thing done to analyze our situation was to analyze the outstanding deviations waiting to be processed. We demonstrated that deviations were piling up in the system, some of them having been waiting for more than thirty days, thus penalizing product release and generating significant disorganization. An action in Breakthrough Kaizen mode allowed us to work on the organization of deviation flows (Fig. 6). Thus, after having processed the “old” deviations, the process analysis taught us that deviation management is underpinned by two critical steps:
- Referral of information to the team that has to handle the deviation.
- The investigation (including impact analysis and determination of root causes).
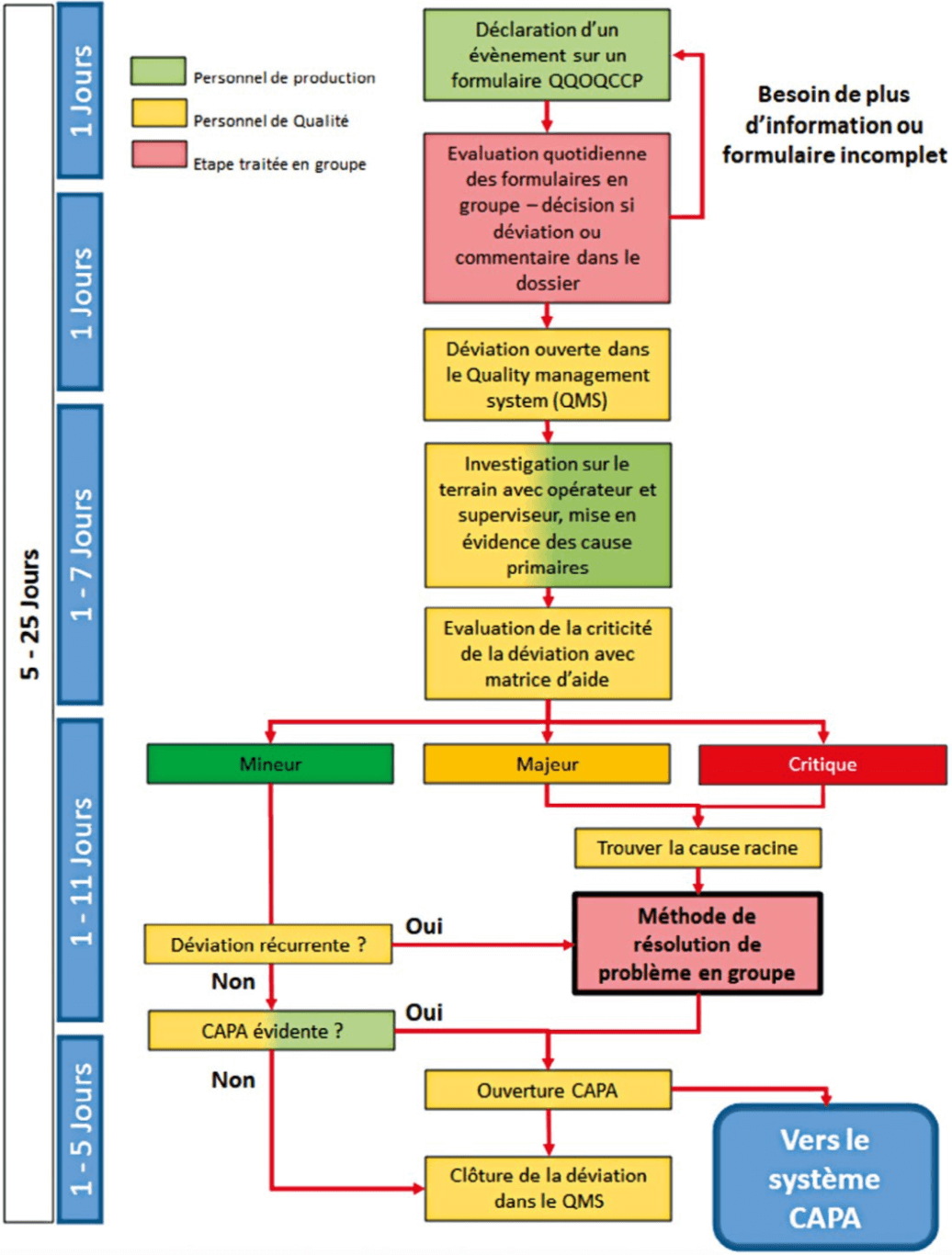
4.2.2 Reporting of information
One of the major irritants of the deviation process was the reliability of information reporting and the time involved in receiving this information. It was not uncommon to discover an anomaly 5 days after it occurred and for it to take up to 20 days after this to be in possession of all the information needed to handle the anomaly. This time frame leads inevitably to loss of information that is crucial for successful completion of operations.
To overcome this difficulty, two things were put in place:
- A form, completed by the operatives, on the ground, at the time of the deviation, reviewed by supervisors to be reviewed by QA.
- An expansion of the ritual described in 4.1.2: during the ritual supervisor and QA exchange information on malfunctions that occurred the previous day and decide whether they should be described as deviations. All new deviations were incorporated into the new visual management system.
In three months, the “pending” level was divided by 3 and deviation handling time fell from 60 days to less than 25 calendar days.
We applied the same pattern to deviations as to the batch record review flow, namely definition of incoming and outgoing flows combined with daily visual management in the form of a process associated with precise time frames for each action.
4.2.3 The investigation
Once the right information has been referred at the right time, half of the work has already been completed. To ensure uniformity of investigations and decisions, investigation forms and decision aid matrices were created for all steps.
As shown on the diagram in 4.2.1 maximum time frames and players were defined for each step.
The points handled in a group (in red on the diagram), adjusted when the deviation requires it, are essential to the proper operation of this system and that of the CAPA system. In effect, this allows the involvement of other professions and so gives meaning to the actions that they will themselves define and carry out later.
Roles are divided between members of production and quality assurance staff: Production is the owner of the production process and is responsible for making it reliable and robust, Quality assurance has a methodological role in the handling of deviations and is responsible for ensuring traceability. The consequence is an organization where it is the operatives that handle the deviation technically using the DMAIC method to define the problem, find its root causes and implement technical solutions. Quality Assurance has the task of ensuring that the methodology meets GMP requirements and implements actions associated with traceability and deviations.
In conclusion, application of the method to deviations has allowed:
- compliance with deviation handling time frames;
- handling of deviations according to the FIFO method;
- the reduction of deviation handling time;
- the reduction of the time frame for opening a deviation after the incident;
- improved efficiency of the deviation process saving time for the pharmacist and production managers.
4.3 Release activity
For a record to be released, the results of the analyses must be validated, the record reviewed and the impact of any malfunctions evaluated.
Before the activities of record review and malfunctions management were brought under control, availability dates were constantly disrupted by emergencies associated with non-finalized investigations. As soon as the flows were brought under control, availability dates became more predictable and reliable.
With the main flow under control, it was then possible to focus on reducing the recurrence of deviations and continuous improvement by means of the CAPA system.
4.4 Management of CAPA
The final cornerstone of QA operational activity, the CAPA management process represents a “spillway” for all processes generating actions such as deviations, out of specifications, audits, annual product quality reviews, customer complaints. CAPAs are at the heart of the quality assurance system.
In our case CAPA management was not efficient for the following reasons:
• Lack of meaning
• Lack of short-term benefit
• Lack of system appropriation
The consequences were a gradual reduction in the number of actions carried out and clogging of the system making it ineffective.
As with the deviation management process, a phase of awareness-raising and empowerment was necessary. QA is responsible for the system, production managers are responsible for carrying out actions within the allotted time frames.
Line management positioning was still more important in this phase as the CAPA process is outside the production flow (see diagram 3.1). It is nevertheless necessary as it will guarantee the sustainability and robustness of the entire QA system. In fact, if we do not work on the recurrence of anomalies and the efficacy of actions that are introduced, the quality system will quickly self-saturate as new faults will appear.
The goal is to focus on recurring anomalies as if the fault returns it is because the root cause was not identified or handled correctly.
A 30-minute ritual conducted at two-week intervals was created to ensure that actions in progress were not encountering problems and that the stated time frames would indeed be observed. It is often necessary to carry out several actions (action plan) to get to the root cause of an anomaly. And a delay in carrying out an action may have consequences for the whole action plan. Visual management makes the invisible visible and ensures that actions are progressing correctly.
To maintain an effective system, the number of action plans opened in relation to the number of deviations opened, and the number of actions opened in relation to the number of actions closed is challenged to avoid clogging the system.
Operational players in production and maintenance are also consulted on two crucial subjects when CAPA are opened:
- At the end of deviation investigations, to find out their opinions on the actions/tests to be conducted on the root cause identified
- After the decision on actions, to determine the time frames acceptable for their performance
An impact/difficulty matrix and another on when to open a CAPA plan to aid decision were defined to standardize practices.
These last points were only possible through control of primary flow management and malfunctions management. CAPA efficacy is monitored via a reduction in recurrent deviations and an improvement in key performance indicators.
4.5 Example of deviation management

It was decided that when a deviation is demonstrated in a management ritual (see 4.1.2), it is handled by the DMAIC method, a multidisciplinary team meets. It is led by a person with Lean certification who ensures the DMAIC process is correctly observed. An internal standard which sets the successive steps is used.

The investigation is conducted searching for potential root causes, then analyses are carried out on the ground.
Tests are decided on for the following week, this constitutes the first CAPA exercise. There is no administrative burden and everything is entered directly into our quality management system (QMS) during the DMAIC activity without spending too much time on it.
The tests allowed us to find the attributable root cause, then adjustments to the equipment were decided on, this constitutes the second CAPA exercise.
Performance monitoring in rituals, allows a check to be made that the CAPAs decided on are effective and that the deviation is no longer being reproduced.
This type of DMAIC is conducted at a relatively high frequency on issues that significantly impact production and quality performance processes (deviations).
Conclusion
More than the establishment of a CAPA system, this article describes the correct operation of an operational quality assurance system which forms an integral part of the production system.
The Lean approach combined with the quality approach described in GMP has given meaning to all production unit players. They worked together to improve their everyday practice going beyond the boundaries of their area. Lean as well as quality stopped being an insensitive mechanism, they were applied for the purpose of accomplishing a goal that was clear to everyone: the reduction of production malfunctions to increase production capacity (right first time).
The involvement of management in driving this movement was essential as independently, no department had enough sway over others to initiate this approach.
Knowledge and stabilization of the different processes making up the product release flow are essential to put in place a continuous improvement initiative.
The positive consequences of installing an effective quality assurance system are multiple:
- Productivity increases, boosted by a reduction in technical problems and an increase in the number of compliant products.
- Compliance improves through actions to bring about compliance and through investigations leading effectively to the root causes.
- Availability times are increasingly short and reliable supported by an effective organization.
- The number of stock-outs has been greatly reduced.
The production/release cycle is repeated and improves each time. Processes are increasingly robust and benefit from feedback formalized by the CAPA launched during the previous cycle.
Continuous improvement is a central pillar both of Lean manufacturing and GMP. The rules described in GMP are often perceived as restrictions on performance while Lean principles are considered contrary to GMP principles. In reality, when we look at the founding principles and objectives in their entirety, GMP and Lean have the same goal: to improve process efficiency and product quality while taking account of constraints. Quality and Lean work most effectively together.
Partager l’article


Chakyr Djane – Vetoquinol
Chakyr DJANE, membre de l’UL6S, est actuellement responsable groupe excellence opérationnelle pour l’industriel chez Vetoquinol. Selon une approche systémique, il déploie au niveau mondial une culture de la performance qui va de la définition de la stratégie à la mise en œuvre sur le terrain, en travaillant autant les méthodes que les organisations et les comportements. Dans l’industrie pharmaceutique, mécanique et photographique, il a aussi travaillé à l’amélioration des performances opérationnelles comme responsable Assurance Qualité et responsable Supply-Chain. Chakyr est ingénieur des industries chimiques (ENSIC), Black Belt Lean, CPIM et coach en intelligence émotionnelle.

Yann Duban – Delpharm Huningue
Responsable d’unité de production – Pharmacien industriel Après 9 années à travailler dans l’industrie pharmaceutique sur des fonctions allant de la qualité opérationnelle au management de site de production à l’étranger, Yann est un fervent défenseur de l’idée que la qualité et la production sont indissociables. Les services d’assurance qualité amèneront de la productivité aux ateliers et les services de production amèneront de l’efficience aux services d’assurance qualité. De plus avec les outils du Lean Manufacturing, la synergie de cet ensemble peut amener à des résultats exceptionnels

Paul Adrien Mathon – Académie de la Qualité Efficace
Depuis 8 ans, Paul-Adrien, Pharmacien Industriel, a occupé des postes proches du terrain en qualité comme en production, dans différents pays et travaille aujourd’hui comme consultant. Cette diversité lui a permis de constater le manque d’efficacité, la récurrence et la similarité des problèmes dans les sites. Il déploie aujourd’hui des approches innovantes pour éliminer ces problématiques et ainsi donner plus de temps aux équipes Qualité pour faire ce qui compte vraiment : accompagner le terrain et faire de l’amélioration continue.
paul.adrien.mathon@gmail.com
