
Summary
- Current state & trends for Single-Use technologies implementation in the Biopharmaceutical Industry
- Continuous Processing. Performance Enhancements for Perfusion Applications in 50L to 500L Single-Use Bioreactors: A Technical Comparison of Performance Characterization, Cell Culture & Scale-Up Modeling
- Implementation of Single-Use in Drug Substance filling before transportation: Product Development case study
- Single use connection technologies: current situation and trends
- Extractables and Leachables from SUS - aspects beyond Extractables Measurement & standardization
- Toxicological evaluation of extractables and leachables associated with the use of Single Use Systems (SUS)
Regulatory bodies expect pharmaceutical companies to guarantee the effectiveness and safety of the medicinal products that are dispensed to patients. Since the use of Single Use Systems, also called disposables, has become a common practice in manufacturing processes, industrialists must provide proof that no toxic substances derived from the materials used in these systems migrate into the finished product.

The multiplicity of materials, their composition and conditions of use make evaluation difficult. This is why an evaluation approach structured according to risk enables prioritization of the studies to be conducted to make their use safe.
Studies of interactions between the primary packaging and the medicinal product are required as part of the submission of marketing authorization dossiers, so as to provide proof that potentially toxic leachables derived from the container, whatever its nature, will not be administered to the patient. Unlike glass containers, which are in most cases relatively neutral in relation to products, plastic (syringes, vials, blisters) or elastomer components (caps, piston gaskets), in direct contact with the products, are less inert and present more risk of leaching. Different European (1) and American(2) regulatory guidelines define the information that needs to be produced for qualification of packaging items. In addition to these texts, the pharmacopeias specify the characteristics of the different commonly used polymers and the tests to be performed to justify their product compatibility.
Some polymer components used during the manufacturing process are also the subject of systematic studies of leachables when no subsequent step enables the removal of potential impurities generated by degradation of the different component parts of the device. This is the case, for example, with filters used for the final sterilization of injectable products for which qualification must provide proof that they are not a source of particulate or chemical contamination.
To provide more flexibility in production and limit risk of cross contamination, some companies have opted to use Single Use systems (SUS) in the manufacture and storage of products or intermediates at different stages of the process. The replacement of stainless steel or glass equipment by equipment mainly composed of polymers is not without impact on the safety of the finished product. In fact, the conditions in which these components are used (temperature, pressure, pH,…) can cause accelerated degradation of the component materials and introduce possibly toxic impurities which would not necessarily be removed during the purification steps.
The diversity of polymers, additives (antioxidants, plasticizers, catalysts) and solvents involved in the composition of disposables and the accumulation of potential impurities throughout the process are factors which must be taken into account in evaluating the risk associated with their presence in the medicinal product. The multiplicity of potential sources of contaminants makes identification and quantification of impurities leached out from the polymer devices used in manufacture difficult. In the absence of regulatory texts specific to the use of SUS, different associations have been created to address the problem associated with extractables and leachables (e.g.: PQRI, BPOG, PDA, BPSA, the A3P SUS CIG, the magazine La Vague…) and to provide methodological and scientific support for the evaluation of disposables.
In application of the process described in ICH Q9(3), a risk assessment should be conducted for each process step for which a SUS is used. This estimation of risk will take account of the sensitivity of the material to extraction (type of polymer), the extraction capacity of the product (solid, liquid, nature and composition of the content, …) and the conditions of use of the component (temperature, duration of contact, volume/surface ratio, pH…) (4 – 5 – 6). The aim of this phase is clearly to categorize the disposables in accordance with the real risk of leaching so as to adjust actions to bring this issue under control and in particular to prioritize the studies of extractables and leachables to be conducted. The analysis process must be adapted to the level of risk and should allow clear rulings to be made on the safety of the systems used. By way of example, the different phases of evaluation for components posing a moderate risk are described in the diagram.
As a reminder, studies of extractables are not required for components posing a low risk. The sole expectation is compliance with the European Pharmacopoeia tests and USP Class VI biological tests (7-8).
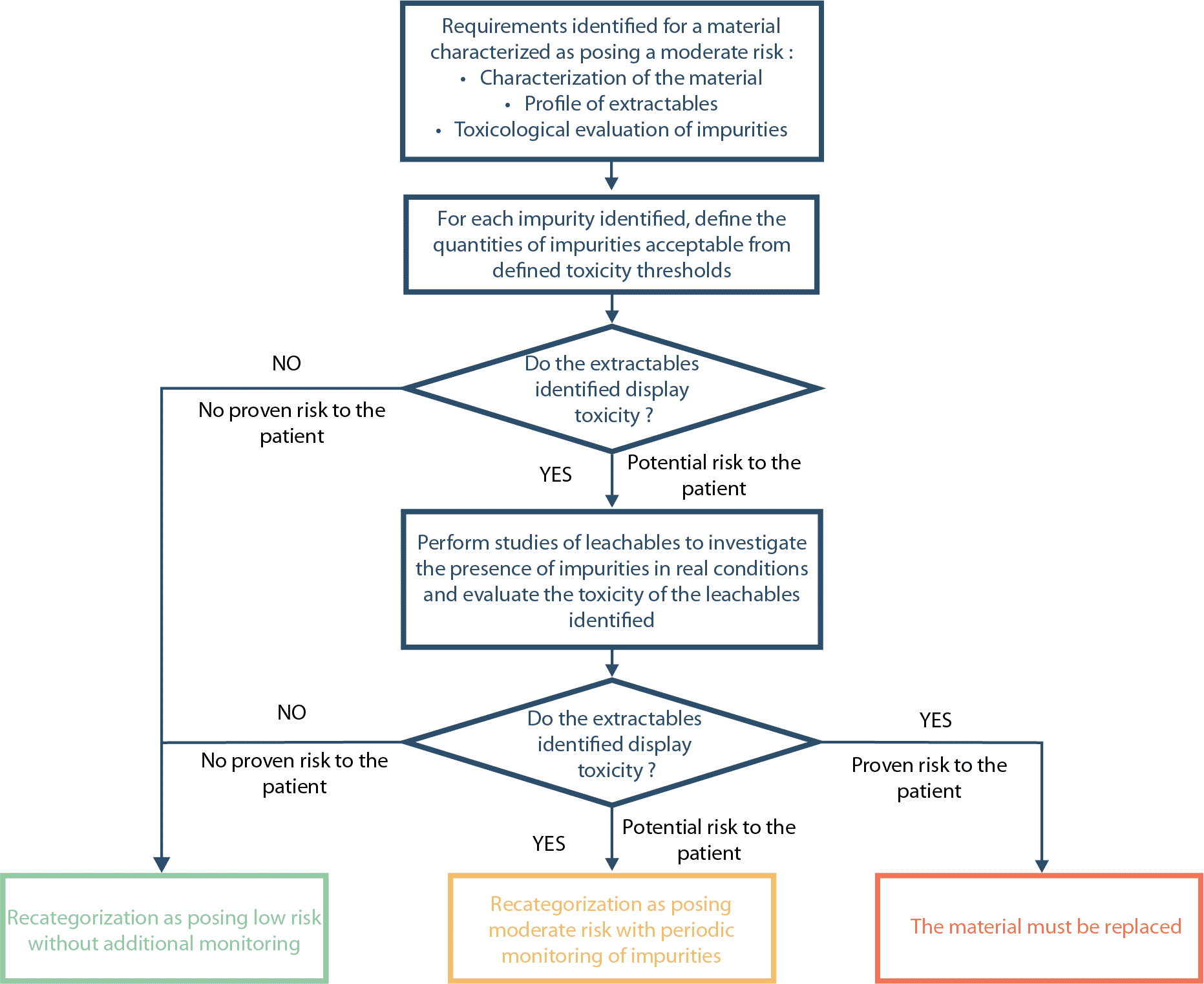
For components posing a moderate or high risk, the identification and quantification of impurities derived from the polymer in contact with the product are necessary so as to evaluate the risk associated with their toxicity(9). Information on the potential impurities derived from the components can come either from supplier validation dossiers or studies of extractables conducted in simulation conditions that are aggressive to the material. By way of example, the BioPhorum Operations Group (BPOG) proposes a test strategy to be followed for the study of extractables, specifying the model solvents to be used and the operating conditions to be followed depending on the use of SUS(10). If the extractables identified present toxicity, studies of leachables must be conducted to possibly demonstrate that this toxic extractable is not, in fact, present in the finished product.
An initial selection of substances presenting a risk can and should be made from the volatile substances (solvents, synthesis residues, monomers), semi-volatile substances (additives, degradation products), and non-volatile substances (mainly organic, antioxidants, fatty acids, stabilizers) and elemental impurities (metals) identified, on the basis of safety data, so as to only consider those that present a risk to the patient. The selection criteria are defined depending on their nature and are explained in the following paragraphs.
ICH Q3D(11) stipulates those elemental impurities which must be included in the analysis (Class 1 and Class 2B toxic substances), those which have proven toxicity which must be considered if they are added intentionally (Class 2B) and those which display toxicity depending on the route of administration (Class 3). Class 4 elemental impurities do not display any toxicity and in this case will be excluded from the analysis. The guide also specifies the permitted doses of elements (PDE – Permitted Daily Exposure) which allow the calculation of the limit concentrations acceptable in the final product.

An impurity detected in the finished product at a level of 30% below the PDE is considered negligible and ICH Q3D(11) specifies that it does not require specific monitoring. For levels ranging from 30% to 90% of the PDE, in view of the potential variability of the concentration of leachables, regular periodic tests of finished product batches can be set up to monitor acceptable thresholds and confirm that these have not been exceeded. It is only at the end of this monitoring stage and if thresholds have never been exceeded that regular monitoring can be abandoned and that the risk can be recategorized as low. If the assays show values around or higher than the PDE, the potential risk to the patient should be re-assessed on a case by case basis. If the risk is proven, methods to reduce the impurity must be sought, and, in their absence, the component material of the disposable must be replaced.
This approach is also applicable for residual solvents which ICH Q3C(R5)(12) classifies according to toxicity and defines their PDE. The limit concentration of solvent is determined according to the formula:
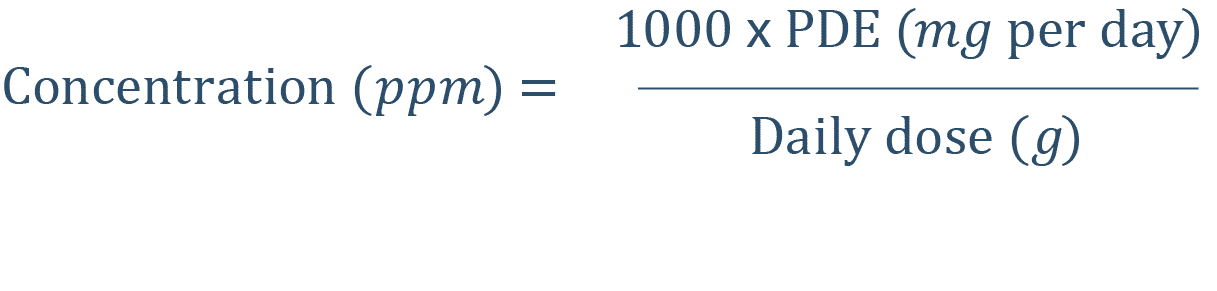
The risk assessment rules applied to elemental impurities can also be applied to residual solvents.
With respect to extractables or leachables which are not covered by the ICH guidelines, depending on their concentration, a toxicological assessment is necessary to rule on whether these compounds present a real risk to the patient and to define the thresholds acceptable in the medicinal product. The toxicological analysis should not be limited to tests of acute, chronic and subchronic toxicity (LD50 relative to the route of administration of the medicinal product), of course indicators of the class of the substance under consideration, but data from the literature concerning the carcinogenic, mutagenic and reproductive toxicity potential should also be included. Depending on the typology of the toxicological risk identified and the mode of administration, two thresholds are possible. For impurities with carcinogenic or mutagenic potential, the PQRI(13) has set the toxicity threshold (SCT – Safety Concern Threshold) at 0.15 μg/day for the nasal or inhaled route and for highly carcinogenic substances and at 1.5 μg/day for the other routes of administration. For substances which do not present a proven oncogenic or mutagenic risk, the SCT has been defined independently of the pharmaceutical form, by default, at 1.5 μg/day for each impurity to take account of the compounds for which no toxicological information was available. These values were established by the analysis of scientific data derived from toxicological databases such as HSDB (Hazardous Substances Data Bank) and IRIS (Integrated Risk Information System). In view of the safety factor applied to SCT, leachables present below this threshold do not require additional assessment. To select the extractables or leachables to be included in the analysis, an analytical threshold in μg/g of container (AET – Analytical Evaluation Threshold) is defined according to the calculation:

In the event that the container is an assembly of different parts, the weight of the whole device must be considered in calculating the AET. A corrective factor can be applied to take account of the variability of the sample processing method and the uncertainty of the test method. The early determination of the AET on the basis of supplier data or information in the literature is useful as the identification and quantification of major impurities detected in studies of extractables and leachables are not required below this threshold.
The risk analyses implemented to categorize risks clearly allow identification of the components which require evaluation to secure their use. In view of the difficulty in obtaining reliable or representative information on products from suppliers, the analysis should allow definition of the studies of extractables and leachables to be conducted to ensure the absence of toxic chemical contaminants derived from materials in contact with the product.
The toxicological assessment of the impurities identified is an essential step in the analysis in order to characterize the risk and to define their acceptable thresholds in the medicinal product and to allow an appropriate monitoring strategy to be introduced.
As with primary packaging items, it is clear that any change in the composition of polymers that are constituents of SUS or their manufacturing process must be managed as part of change control and be the subject of a risk review incorporating new studies and an analysis of the toxicological data of the substances detected.
Share article


Roland OLLIVIER – AKTEHOM
roland.ollivier@aktehom.com
References
(1) Guideline on plastic immediate packaging materials CPMP/QWP/4359/03 of May 29th, 2005
(2) Guidance for Industry et Code of federal register 21CFR – Container closure systems for packaging human drugs and biologics (1999)
(3) ICH Q9: Quality Risk Management. US Fed. Reg. 71(106) 2006
(4) Bennan J. et al, BioPharm International December 2002, 22-34; Evaluation of Extractables from Product-Contact Surfaces
(5) BPOG Best Practices Guide for Evaluating Leachables Risk in Biopharmaceutical Single-Use Systems
(6) The 2010 BPSA Recommendations for Testing and Evaluation of Extractables from Single-Use Process Equipment
(7) USP <87> Biological Reactivity, In Vitro
(8) USP <88> Biological Reactivity Tests, In Vivo
(9) Brochard T.H. et al, Regulatory Toxicology and Pharmacology; 2016 Nov; 81:201-211; Assessing safety of extractables from materials and leachables in pharmaceuticals and biologics – Current challenges and approaches
(10) Ding W. et al, Pharmaceutical engineering 34, 1 – 11, 2014; Standard extraction protocol for Single-Use systems in biopharmaceutical manufacturing
(11) ICH Q3D(R1) – Guideline for elemental impurities – 2018 (Step 2)
(12) ICH Q3C(R6) – Impurities: Guideline for residual solvents – 2016
(13) PQRI Safety Thresholds and Best Practices for Extractables and Leachables in orally Inhaled and Nasal Drug Products – September 2006
Glossary
AET: Analytical Evaluation Threshold
BPOG: BioPhorum Operations Group
BPSA: Bio-process Systems Alliance
HSDB: Hazardous Substances Data Bank
IRIS: Integrated Risk Information System
PDA: Parenteral Drug Association
PDE: Permitted Daily Exposure
PQRI: Product Quality Research Institute
SCT: Safety Concern Threshold
