


Sommaire
- Produits Combinés Injectables. Enjeux et challenges pour les industriels.
- Extension des délais ou non, ce que les fabricants de Classe I doivent retenir.
- Les nouveaux défis des médicaments injectables.
- L’investigation clinique des dispositifs médicaux combinés dans le cadre du nouveau règlement (UE) 2017/745 (RDM).
- Apport de la caractérisation physico-chimique des matériaux constitutifs des dispositifs médicaux pour une rationalisation de leur évaluation biologique.
- Piloter la performance des validations de nettoyage : un enjeu industriel fort.
- Validation des procédés de nettoyage : pourquoi et comment valider les méthodes analytiques et de prélèvements associées.
- Maintenance des équipements en acier inoxydable dans un environnement de fabrication conforme aux BPF : une approche basée sur les risques.
- Single Use Systems vs Re-Usable Stainless-Steel Equipment. Compliance & Quality Perspective.
Apport de la caractérisation physico-chimique des matériaux constitutifs des dispositifs médicaux pour une rationalisation de leur évaluation biologique.
La sécurité biologique des dispositifs médicaux est une exigence réglementaire. Elle intégrée dans le plan de gestion des risques global, impliquant notamment l’évaluation du rapport risque biologique/ bénéfice clinique. Selon la nouvelle version de la norme ISO 10993 encadrant cette évaluation, la caractérisation physico- chimique du dispositif médical, en première intention, s’y avère cruciale.

La protection des patients au regard des risques biologiques potentiels, inhérents à l’utilisation d’un dispositif médical (DM) est une exigence réglementaire inscrite dans les directives européennes ou dans le règlement européen aujourd’hui (MDR 2017/745). Elle est, notamment appréhendée, en première intention, par l’évaluation de sa biocompatibilité. Cet aspect a toujours été l’un des sujets majeurs du développement des DM et fait partie d’une démarche globale de la sécurité biologique qui, elle-même est intégrée au plan de gestion de risque, qui est un processus continu devant être mené par les fabricants de DM, au cours duquel ils doivent identifier, estimer et évaluer les risques biologiques, mais également évaluer le rapport risque biologique global au regard du bénéfice clinique.
Lors de l’évaluation biologique, l’intégralité du cycle de vie du DM, depuis la phase de conception, jusqu’à l’emballage final est passée en revue car, l’origine des risques biologiques provient de la présence des composés de basse masse molaire présents dans les matériaux à tous les niveaux du DM. La composition de ces composés est complexe, en raison de leurs origines multiples. Ils peuvent provenir des procédés de synthèse qui impliquent des résidus, mais également processus de transformation qui nécessitent l’ajout d’adjuvants, mais aussi, des conditionnements, des méthodes de stérilisation, des conditions de stockage et de transport qui génèrent des composés néoformés qui constituent de nouvelles entités pouvant en outre, interagir entre elles et avec les composés originellement présents ou ajoutés au matériaux et au DM. Ainsi la multiplicité des sources potentielles de ces contaminants, rend difficile la caractérisation et la quantification de ces extractibles qui peuvent potentiellement être relargués par le DM dans les milieux biologiques et/ou les formulations médicamenteuses et dont la présence ne peut être appréhendé que par des investigations analytiques appropriées fondées sur la maîtrise des outils et la robustesse des méthodes ainsi que la connaissance de l’état de l’art sur le comportement des matériaux sous l’effet de ces processus de modification et de vieillissement. L’évaluation du risque biologique doit donc porter sur le DM produit fini, ayant subi toute les étapes de sa fabrication à son conditionnement sans oublier, le cas échéant son stockage et l’impact du processus de distribution. Dans ce contexte, la collecte des informations relatives aux caractéristiques chimiques physiques et mécaniques, représente l’étape initiale et apparait comme cruciale dans le processus de caractérisation physico-chimique du matériau qui lui-même, est un prérequis fondamental pour l’évaluation biologique et pour celle des risques biologiques, qui sans ces apports préalables, elles perdront de leur efficience et de leur efficacité.
D’une manière générale, deux catégories de risques devront être appréhendés, il y a les risques généraux correspondant à ceux liés aux procédés de fabrication, aux matériaux de composition des DM et des conditionnements, et par conséquent ceux liés aux substances extractibles, mais également ceux liés aux interactions DM/DM; DM/Accessoires et DM/Médicaments sans oublier ceux liés aux substances issues la non stabilité et au vieillissement, et les risques spécifiques liés aux substances CMR, perturbateur endocriniens, phtalates, nanomatériaux, substances d’origines animales (non viable) et relevant de la sécurité virale. Ainsi, l’évaluation des risques biologiques doit être menée à chaque étape de la fabrication jusqu’à la distribution et le stockage afin de déterminer l’acceptabilité ou non du risque résiduel.
1. Référentiels réglementaires et normatifs Les autorités compétentes attendent des fabricants et des opérateurs économiques associés de garantir, notamment la sécurité d’utilisation des DM mis à disposition des utilisateurs. C’est en partie l’objectif principal des normes de la série ISO 10993. Celles-ci offrent un ensemble d’outils à la fois stratégiques, techniques et scientifiques permettant au fabricant de s’assurer de l’absence de tout risque biologique pour le patient et/ou l’utilisateur d’un DM. Cette série de normes représente une vingtaine de normes que nous avons rassemblées par groupe selon leur spécificité. Le tableau suivant regroupe l’ensemble de ces textes au regard de leur champ d’application. Le document cadre (ISO 10993-1) permet de comprendre les mécanismes de base de la réponse tissulaire, il tend, en outre, vers la réduction, autant que possible, du nombre des expositions animales et privilégie, lorsque celui-ci est pertinent, le modèle in vitro, associé à une évaluation physico-chimique approfondie et justifiée. |  |
Dans ce document, outre la présentation des aspects généraux de l’évaluation, une attention particulière est portée à l’évaluation de toutes les données existantes, à l’identification des manques sur les données disponibles et à l’identification des données supplémentaires nécessaires se rapportant aux matériaux constitutifs des DM mais également ceux utilisés dans leurs processus de fabrication. De ce fait, les données fournisseurs sont fondamentales pour l’évaluation biologique. La demande formulée par les fabricants de DM auprès de leurs fournisseurs, doit être explicite et en accord avec les exigences de la norme. Cela nécessite, toutefois, un travail de fond sur le texte en question.
Bien qu’elle ait été révisée, la version 2018 [ISO 10993-1 (2018)] ne modifie pas fondamentalement le processus d’évaluation biologique, mais clarifie certains points et renforce tout particulièrement la nécessité de réaliser une caractérisation physico-chimique qui est positionnée comme incontournable pour l’évaluation du risque biologique. Ce document apporte en outre, des définitions supplémentaires, comme celles relatives au contact direct, au contact indirect et au contact transitoire. Il est précisé que ce dernier est qualifié comme tel s’il est inférieur à une minute, tenant compte du temps cumulé. Ce type de DM ne nécessite pas la réalisation des tests de biocompatibilité. Néanmoins, les enduits, les lubrifiants ou tous autres adjuvants utilisés dans leur processus de fabrication, peuvent rester en contact avec les tissus même après utilisation du DM. Ainsi, dans ce contexte, ces produits impliquent une attention particulière quant à leur devenir dans l’organisme et peuvent, de ce fait, nécessiter une évaluation de leur biocompatibilité.
2. Démarche scientifique
Au regard de l’ensemble de ces éléments et de ces considérations, permettant d’aborder l’évaluation biologique, il en ressort que ce processus est basé sur une analyse de risques du DM produit fini, tenant compte des risques relatifs aux matériaux constitutifs des DM et des conditionnements primaires, associés à leurs processus de fabrication. L’analyse est donc fondée sur les données collectées se rapportant aux propriétés chimiques, physiques et mécaniques des matériaux constitutifs du DM (produit fini). En effet, les matériaux entrant dans la fabrication des DM doivent répondre au concept de biomatériau. Cela implique sa conformité à des spécifications, sa biocompatibilité et sa stabilité dans les conditions de stockage et d’usage. En outre, les informations collectées concernant les fiches techniques… sont des prérequis fondamentaux pour mener la caractérisation physico- chimique et par conséquent pour prédire la réponse biologique des matériaux entrant dans la composition des DM. Ainsi le document cadre traitant de la caractérisation des matériaux constitutifs des DM, de leurs emballages et de leurs substances extractibles et relargables, est la norme ISO 10993-18. Elle est destinée aux fabricants de DM mais également aux fournisseurs de matériaux et conditionnement. Les exigences spécifiées dans ce document visent à collecter des données relatives à l’identification des matériaux constitutifs du DM, à la détermination qualitative et quantitative des substances chimiques présentes dans ces matériaux et celles utilisées dans les processus de fabrication du DM mais aussi à celles correspondantes à leur potentiel de diffusion à travers les matrices de matériaux et celui de leur relargage dans les conditions d’usage du DM.
Ce document définit, de ce fait un cadre pour l’identification des caractéristiques chimiques liées à la composition du DM. Les données collectées associés à la documentation fournisseur comme les fiches sécurité mais également les certificats de conformité à une monographie, notamment de la pharmacopée européenne qui est centrée autour des propriétés physico-chimiques des matériaux et leur composition, constitue le meilleur moyen de documenter la composition d’un matériau. Ces propos sont clairement énoncés dans le paragraphe B.4 de l’ISO 10993-18 (2020) ou il est mentionné que si ladite norme spécifie un matériau ou une catégorie de matériaux et si elle établit des limites exhaustives, spécifiques, générales ou absolues, pour des substances chimiques présentes dans les matériaux, alors l’indication de la référence de cette norme peut suffire pour la caractérisation du matériau. Ainsi, les monographies de la pharmacopée européenne, relatives aux matériaux répondent parfaitement à ce cas de figure et qui plus est, elles font partie des normes harmonisées. Par ailleurs, il est également dit dans ce paragraphe, que les données de sécurité générées après l’utilisation d’un matériau dans un environnement clinique, peuvent également être considérées dans ce processus de caractérisation physico- chimique. L’ensemble des données collectées constituent les données d’entrées du processus d’évaluation et de maitrise des risques biologiques des DM au cours duquel la norme prévoit de considérer toutes les substances chimiques contenues dans un DM.
De ce fait, il est important, me semble-t-il, de bien comprendre ce qu’est un matériau et plus particulièrement un matériau plastique qui est, dans ce contexte médical, qualifié comme biomatériau et le plus complexe des biomatériaux en termes de caractérisation. En effet, Il est formulé à partir d’un ou plusieurs polymères contenant des résidus de synthèse, auxquels sont ajoutés des adjuvants pour conférer au matériau des propriétés technologiques particulières ou pour le protéger de la dégradation. En outre, pour certains usages, les DM nécessitent d’être stérilisés. L’ensemble de ces processus (synthèse, formulation, fabrication, stérilisation) conduisent à la genèse de substance chimiques, non identifiées ou partiellement identifiées, présentes en quantités variables, susceptibles d’être cédés par les matériaux ou le DM à partir de la surface de contact. Ce mélange complexe est appelé extractible car il peut être extrait dans des conditions agressives, de température, d’agitation, de temps et de polarité de solvant d’extraction. Quant aux relargables, ils font parti de ces extractibles pouvant migrer dans les milieux de contact, soit directement dans le milieu biologique soit indirectement à partir des formulations médicamenteuses administrées, dans les conditions revendiquées par le fabricant, pour l’usage du DM.
Ce contexte complique fortement les analyses qualitatives et quantitatives de ces substances, d’autant plus que ces mélanges évoluent en fonction des conditions de traitement, du stockage et d’utilisation qui génèrent des composés qui en plus peuvent se recombiner entre eux. Compte tenu de la complexité de ces mélanges d’extractibles et de relargables, la nécessité de déployer une stratégie scientifique justifiée s’avère cruciale. Elle doit associer la collecte de données (fournisseurs, état de l’art…) et une approche analytique appropriée justifiant le choix des techniques analytiques qui lui sont associées. Ces méthodes se déclinent en deux catégories celles destinées à extraire les substances de leur matrice polymère et celles nécessaires à leur détermination qualitative et quantitative.
3. Méthodologie analytique
Les méthodes d’extraction sont destinées à produire un profil d’extractibles égal ou supérieur à celui que génèreraient les conditions d’usage du DM, sans pour autant que la méthode ne crée de nouveau produits de dégradation. La norme ISO 10993-18 nouvelle version définit trois approches : l’extraction simulée, dans ce cas le milieu d’extraction mime celui de l’usage, l’extraction exagérée, qui a pour objectif de forcer considérablement la libération des extractibles et l’extraction exhaustive qui consiste en une répétition des étapes d’extraction. Il est préconisé dans la norme, de justifier le choix de la méthode d’extraction. En outre des recommandations y sont également apportées en fonction du temps de contact du DM avec l’organisme (Tableau 2).
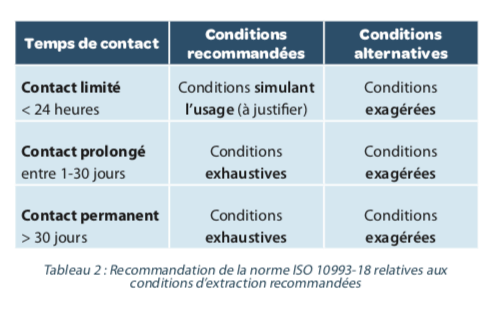
Il est par ailleurs recommandé de démontrer la répétabilité et la reproductibilité de la méthode d’extraction, ce qui permettra de justifier le nombre d’extractions à réaliser.
Quant à la méthodologie analytique à déployer pour l’analyse des extractibles. Il est préconisé dans la norme que les méthodes qualitatives développées puissent permettre d’identifier ces composés et qu’elles soient quantitatives. Il est admis que pour les composés organiques volatils des méthodes par chromatographie gaz–liquide couplées avec un dispositif “head–space” pour les extraire du matériau sont les plus appropriées. Alors que pour les composés non volatils, une analyse par chromatographie liquide serait plus adaptée. Les extractibles atomiques de type métaux et non métaux sont, quant à eux spécifiquement déterminés qualitativement et quantitativement par des méthodes de spectroscopie atomique de type SAA et ICP-MS.
C’est ainsi que toutes les méthodes décrites dans la norme ne sont pas obligatoires, si au regard de la connaissance de l’état de l’art et de la documentation fournisseurs certains composés peuvent être exclus du profil des extractibles. Néanmoins, toute stratégie analytique déployée doit être justifiée. En outre chaque méthode utilisée doit être validée en termes de spécificité, de limite de détection et de quantification, de précision, d’exactitude, de linéarité et de stabilité, pour que les extractibles obtenus après criblage des solutions d’extraction, puissent être identifiés et dosés.
L’objectif final d’une telle stratégie analytique est de pouvoir considérer l’impact toxicologique de chaque composé identifié comme pouvant être potentiellement relargué dans des conditions d’usage. Cependant, cette identification n’est pas toujours aisée et par voie de conséquence, l’évaluation des risques toxicologiques comme le décrit la norme ISO 10993-17 ne peut être effectuée. Ainsi la notion de seuil d’évaluation analytique (AET) a été introduite dans la norme ISO 10993-18 nouvelle version. Cette démarche consiste à établir un seuil de concentration analytique pour les extractibles et les relargables. Ainsi, seront considérés comme potentiellement toxique et par conséquent entrant dans l’évaluation des risques toxicologiques, tous analytes détectés sur le profil des extractibles et dont la concentration est supérieure à l’AET, à contrario, les autres composés de concentration inférieure à l’AET ne seront pas considérés dans cette évaluation (figure 1). Toutefois, ce seuil (AET) n’est applicable qu’aux composés organiques.
La détermination de l’AET implique deux conditions relatives aux méthodes utilisées. Ainsi, l’AET doit être supérieur ou égal à la limite de détection de la méthode (LOD), cela indiquera sa capabilité à détecter les relargables pertinents. Cette LOD peut être déterminée au moyen d’étalons internes dont les caractéristiques physico-chimiques se rapprocheraient au mieux des composés criblés. En outre si la méthode est appliquée à des fins de quantification l’AET doit également être supérieur ou égal à la limite de quantification (LOQ).
Ainsi, l’AET peut être calculé en incluant comme données d’entrée : la fréquence et la durée d’usage du DM, les conditions d’extraction aboutissant au profil d’extractibles et l’incertitude relative à la méthode analytique.

B = Volume de l’extrait (ml).
C = Exposition clinique au DM = le nombre de DM auquel l’utilisateur est exposé en 1 jour selon la pratique clinique revendiquée.
UF = Coefficient d’incertitude analytique découlant de la variabilité de l’exactitude.
Le degré d’incertitude dépend de la méthode de quantification appliquée. Ainsi :
• Si le dosage est réalisé au moyen d’un étalon interne, la réponse des extractibles est normalisée avec comme hypothèse que l’ensemble des analytes ont une réponse similaire au composé choisi, dans ce cas le degré d’incertitude UF est élevé.
• Si le dosage est réalisé au moyen d’un étalon externe authentique, dans ce cas l’estimation de la concentration des analytes est suffisamment exacte et de ce fait, le degré d’incertitude UF est faible.
Par ailleurs, entre ces deux estimations extrêmes, d’autres stratégies de dosage pourrait conduire à des estimations intermédiaires, comme par exemple, l’application des facteurs de réponse relatifs, extractible versus étalon interne.
Actuellement, aucune préconisation n’est faite pour une valeur spécifique de ce coefficient UF. Il est, toutefois recommandé d’appliquer une approche statistique pour établir et justifier le choix d’une valeur pour ce coefficient.
Ainsi, l’utilisation de ce seuil (AET) permet de justifier le choix d’une évaluation biologique ou non sur chaque extractible ou relargable déterminé dans un échantillon.
Conclusion
Les risques biologiques relatifs à l’utilisation clinique d’un DM sont, dans bien des cas, dus à leurs propriétés de surface. En effet, certaines caractéristiques comme la chimie, la rugosité et/ou l’énergie de surface impactent fortement la biocompatibilité d’un DM et conduisent à des réactions locales. Il n’en demeure pas moins, que des effets systémiques sont souvent cités dans les études précliniques et sont liés à la libération dans le système circulatoire, de composés (relargables) situés à l’interface DM/milieu biologique ou DM/formulation médicamenteuse. La compréhension et l’anticipation de ces risques passent par une connaissance approfondie des matériaux utilisés dans la fabrication des DM. C’est ainsi qu’il est parfaitement admis que le succès d’une évaluation des risques biologiques d’un DM est directement lié à celui de sa caractérisation physico- chimique, impliquant ces propriétés surfaciques, volumiques et sa composition. De ce fait, une telle démarche nécessite obligatoirement la collaboration de spécialistes de la physico-chimie des polymères, des chimistes analystes et des biologistes ayant tous un socle de compétences commun : la connaissance du DM.
Partager l’article

Katia MANERLAX, Lionel TORTOLANO & Najet YAGOUBI – Univ.Paris Saclay, UFR de Pharmacie, Lab. Matériaux & Santé EA 401.
Références
Exudation of additives to the surface of medical devices: impact on biocompatibility in the case of polyurethane used in implantable catheters. J Biomed Mater Res A., Volume104 (12), 2016, p. 2954- 2967
Interaction of intraocular lenses with fibronectin and human lens epithelial cells: Effect of chemical composition and aging. J Biomed Mater Res A., volume 103(12), 2015, p.3843-51
