


Summary
- Injectable Combination Products. Issues and challenges for industry.
- Deadline Extension or Not: Key Points for Class I Manufacturers
- The new challenges of injectable medicines
- Clinical Investigation of Combined Medical Devices Under New Regulation (EU) 2017/745 (MDR)
- Contribution of the physicochemical characterization of the materials constituting medical devices for rationalisation of their biological assessment.
- Steering Cleaning Validation Performance : A Key Industrial Challenge.
- Cleaning Process Validation: Why and How to Validate Analytical Methods and Related Sampling Methods
- A Risk-Based Approach to Stainless Steel Equipment Maintenance in cGMP Manufacturing Environment
- Single Use Systems vs Re-Usable Stainless-Steel Equipment. Compliance & Quality Perspective.
Contribution of the physicochemical characterisation of the constituent materials of medical devices for rationalisation of their biological assessment.
The biological safety of medical devices is a regulatory requirement. It is integrated into the overall risk management plan, notably involving the assessment of the biological risk / clinical benefit ratio. According to the new version of ISO 10993 standard framing this assessment, the physicochemical characterisation of the medical device, in the first intention, turns out to be crucial.

The protection of patients against the potential biological risks inherent in the use of a medical device (MD) is a regulatory requirement enshrined in European directives or European regulation today (MDR 2017/745). It is, in particular, apprehended, in the first line, by the assessment of its biocompatibility. This has always been one of the significant topics of MD development and is part of a comprehensive biological safety approach that is itself integrated into a risk management plan, which is an ongoing process to be conducted by MD manufacturers, during which they must identify, estimate and assess biological risks, but also determine the overall biological risk ratio for clinical benefit.
During the biological assessment, the entire lifecycle of the MD, from the design phase, to the final packaging is reviewed because the origin of the biological risks comes from the presence of the compounds of low molar mass present in materials at all levels of the MD. The composition of these compounds is complicated due to their multiple origins. They can come from synthetic processes which involve residues, but also transformation processes which require the addition of adjuvants, but also, packaging, sterilisation methods, storage and transport conditions which generate neoformed compounds which constitute new entities which can furthermore interact with each other and with the compounds initially present or added to the materials and the MD. Thus the multiplicity of potential sources of these contaminants, makes it challenging to characterise and quantify these extractables, which can potentially be released by MD into biological environments and/or drug formulations and whose presence can only be apprehended by appropriate analytical investigations based on the mastery of tools and robustness of the methods as well as knowledge of state of the art on the behaviour of materials as a result of these processes of modification and ageing. The assessment of the biological risk must, therefore, focus on the finished product MD, having undergone all the stages of its manufacture to its packaging without forgetting, if necessary its storage and the impact of the distribution process. In this context, the collection of information on physical and mechanical chemical characteristics represents the initial stage. It appears to be crucial in the process of physicochemical characterisation of the material, which itself is a fundamental prerequisite for biological and biological risk assessment, which without these prior inputs will lose their efficiency and effectiveness.
In general, two categories of risks will need to be addressed, with the general risks associated with manufacturing processes, MD and packaging composition materials, and, therefore, those related to extractable substances, but also those pertaining to MD/MD interactions; MD/Accessories and MD/Drugs, not to mention those related to substances resulting from non-stability and ageing, and specific risks associated with CMR substances, endocrine disruptors, phthalates, nanomaterials, animal substances (not viable) and viral safety. Thus, the biological risk assessment must be conducted at every stage from manufacturing to distribution and storage to determine whether or not the residual risk is acceptable.
1. Regulatory and normative standards The relevant authorities expect manufacturers and associated economic operators to guarantee, in particular, the safety of the use of MD made available to users. This is partly the main objective of the ISO 10993 series standards. These provide a set of strategic, technical and scientific tools that enable the manufacturer to ensure that there is no biological risk to the patient and/or the user of an MD. This set of standards represents about twenty standards that we have grouped according to their specificity. The following table groups all of these texts in terms of their scope. The framework document (ISO 10993-1) provides an understanding of the underlying mechanisms of tissue response, and it also tends towards reducing, as much as possible, the number of animal exposures and favours, where relevant, the in vitro model, combined with a thorough and justified physicochemical assessment. |  |
In this document, in addition to presenting the general aspects of the assessment, special attention is paid to the assessment of all existing data, the identification of gaps in available data and the identification of the additional data needed relating to the constituent materials of the MD but also those used in their manufacturing processes. As a result, supplier data is fundamental to biological assessment. The request made by MD manufacturers to their suppliers must be explicit and following the requirements of the standard. This, however, requires substantive work on the text in question.
Although revised, the 2018 version [ISO 10993-1 (2018)] does not fundamentally alter the biological assessment process, but clarifies specific points and particularly reinforces the need for physicochemical characterisation that is positioned as essential for biological risk assessment. Although revised, version 2018 [ISO 10993-1 This document also provides additional definitions, such as direct contact, indirect contact and transient contact. It is specified that the latter is qualified as such if it is less than one minute, taking into account the cumulative time. This type of MD does not require biocompatibility testing. However, coatings, lubricants or other additives used in their manufacturing process may remain in contact with tissue even after the MD is used. Thus, in this context, these products involve special attention as to their future in the body and may, therefore, require an assessment of their biocompatibility.
2. Scientific approach
Given all of these elements and considerations, which allow for the approach of biological assessment, it appears that this process is based on a risk analysis of the finished MD product, taking into account the risks related to the constituent materials of the MD and primary packaging, associated with their manufacturing processes. The analysis is, therefore, based on the data collected relating to the chemical, physical and mechanical properties of the constituent materials of the MD (finished product). Indeed, the materials used in the manufacture of the MD must meet the concept of biomaterial. This implies its compliance with specifications, its biocompatibility and its stability in storage and usage conditions. Besides, the information collected regarding the technical sheets is fundamental prerequisites for conducting physicochemical characterisation and, therefore, for predicting the biological response of materials in the composition of the MD. Thus, the framework document dealing with the characterisation of the constituent materials of the MD, their packaging and their extractable and biofuel substances, is ISO 10993-18. It is intended for manufacturers of MD but also for suppliers of materials and packaging. The requirements specified in this document are intended to collect data relating to the identification of the constituent materials of DM, the qualitative and quantitative determination of the chemicals present in these materials and those used in the manufacturing processes of DM but also those corresponding to their potential for diffusion through the matrixes of elements and that of their release in the conditions of the MD’s usage.
The requirements specified in this document are intended to collect data relating to the identification of this document defines, therefore, a framework for the identification of chemical characteristics associated with the composition of the MD. The data collected associated with supplier documentation such as safety cards but also certificates of compliance with a monograph, in particular European pharmacopoeia, which focuses in the physicochemical properties of materials and their composition, is the best way to document the formation of an article. These words are clearly stated in paragraph B.4 of ISO 10993-18 (2020), or it is said that if that standard specifies a material or category of materials and if it establishes comprehensive, specific, general or absolute limits for chemicals present in the materials, then the indication of the reference of that standard may suffice for the characterization of the material. Thus, the monographs of the European Pharmacopoeia, relating to materials, perfectly meet this scenario, and they are part of the harmonised standards. Furthermore, it is also stated in this paragraph that safety data generated after the use of the material in a clinical environment, can also be considered in this process of physicochemical characterisation. All of the data collected are input data from the biological risk assessment and control process for MDs, during which the standard provides for all chemicals contained in an MD.
Therefore, it seems to me that it is essential to understand what a material is and in particular a plastic material which is, in this medical context, qualified as a biomaterial and the most complex of biomaterials in terms of characterisation. Indeed, it is formulated from one or more polymers containing synthetic residues, to which additives are added to give the material unique technological properties or to protect it from degradation. Besides, for specific uses, MDs need to be sterilised. All of these processes (synthesis, formulation, manufacture, sterilisation) lead to the genesis of chemical substances, unidentified or partially identified, present in variable quantities, liable to be transferred by the materials or the MD from the contact surface. This complex mixture is called extractable because it can be extracted under aggressive conditions, temperature, agitation, time and polarity of extraction solvent. As for the leachables, they are part of these extractible that can migrate into contact environments, either directly into the biological environment or indirectly from the drug formulations administered, under the conditions claimed by the manufacturer, for the use of DM.
This context greatly complicates qualitative and quantitative analyses of these substances, especially as these mixtures evolve according to the processing, storage and use conditions that generate compounds that can also recombine among themselves. Given the complexity of these mixtures of extractable and releases, the need to deploy a justified scientific strategy is crucial. It must combine data collection (suppliers, state of the art, etc.) with an appropriate analytical approach justifying the choice of analytical techniques associated with it. These methods come in two categories, those designed to extract substances from their polymer matrix and those necessary for their qualitative and quantitative determination.
3. Analytical methodology
The extraction methods are intended to produce a profile of extractable equal to or higher than that generated by the conditions of use of the MD, without the technique creating new degradation products. The new version of the ISO 10993-18 standard defines three approaches: simulated extraction, in this case, the extraction medium mimics that of use, exaggerated extraction, which aims to considerably force the release of extractable and exhaustive extraction which consists of a repetition of the extraction steps. It is recommended in the standard, to justify the choice of extraction method. Recommendations are also made based on the time the MD has contact with the organization (Table 2).
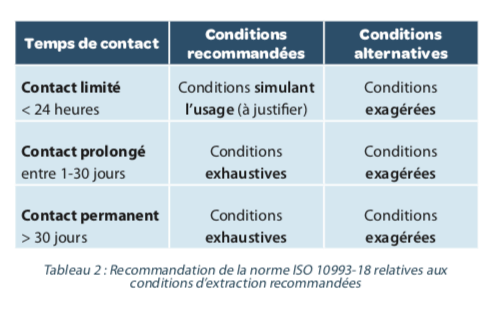
It is also recommended to demonstrate the repeatability and reproducibility of the extraction method, which will justify the number of extractions to be carried out.
As for the analytical methodology is to be deployed for the analysis of extractable. It is recommended in the standard that the qualitative methods developed can make it possible to identify these compounds and that they are quantitative. It is recognised that for volatile organic compounds, gas-liquid chromatography methods coupled with a “head-space” device to extract them from the material are the most suitable. Whereas for non-volatile compounds, an analysis by liquid chromatography would be more suitable. The metal and non-metal type atomic extractable are individually determined qualitatively and quantitatively by atomic spectroscopy methods of the SAA and ICP-MS type.
Thus, not all methods described in the standard are mandatory, if, in terms of knowledge of state of the art and documentation suppliers, specific compounds may be excluded from the profile of extractable. However, any analytical strategy deployed must be justified. Besides, each method used must be validated in terms of specificity, detection and quantification limit, precision, accuracy, linearity and stability, so that extractable obtained after a screening of extraction solutions can be identified and measured.
The ultimate goal of such an analytical strategy is to be able to consider the toxicological impact of each compound identified as potentially released under conditions of use. However, this identification is not always easy and, as a result, toxicological risk assessment, as described in ISO 10993-17, cannot be carried out. Thus the concept of analytical assessment threshold (AET) was introduced in the new version of ISO 10993-18. This approach consists in establishing an analytical concentration threshold for extractables and leachables. Thus, will be considered as potentially toxic and, therefore, entering into the assessment of toxicological risks, all analytes detected on the profile of extractable, and whose concentration is higher than the AET, on the contrary, the other compounds of concentration lower than the AET will not be considered in this assessment (Figure 1). However, this threshold (AET) is only applicable to organic compounds.
Determining the AET involves two conditions relating to the methods used. Thus, ACT must be greater than or equal to the method detection limit (LOD); this will indicate its capability to detect relevant leachables. This LOD can be determined using internal standards whose physicochemical characteristics would best approximate the screened compounds. Besides, if the method is applied for quantification purposes, the AET must also be greater than or equal to the limit of quantification (LOQ).
Thus, the AET can be calculated by including as input data: the frequency and duration of use of DM, the extraction conditions leading to the extractability profile and the uncertainty regarding the analytical method.

B = Volume of extract (ml).
C = Clinical exposure to MD = the number of MD to which the user is exposed in 1 day according to the clinical practice claimed.
UF = Analytical uncertainty coefficient resulting from accuracy variability.
The degree of uncertainty depends on the quantification method applied. Thereby:
• If the dosage is performed using an internal standard, the extractable response is standardized with the assumption that all analytes have a similar response to the chosen compound, in this case, the degree of uncertainty UF is high.
• If the dosage is done using an authentic external standard, in this case, the estimate of the concentration of analytes is sufficiently accurate and, therefore, the degree of uncertainty UF is low.
Furthermore, between these two extreme estimates, other dosing strategies could lead to intermediate estimates, such as the application of relative response factors, extractable versus internal standard.
Currently, no recommendation is made for a specific value of this UF coefficient. It is, however, recommended to apply a statistical approach to establish and justify the choice of a value for this coefficient.
Thus, the use of this threshold (AET) makes it possible to justify the choice of a biological assessment or not on each extractable or releasable determined in a sample.
Conclusion
The biological risks associated with the clinical use of an MD are, in many cases, due to their surface properties. Indeed, specific characteristics such as chemistry, roughness and/or surface energy strongly impact the biocompatibility of a DM and lead to local reactions. The fact remains that systemic effects are often cited in preclinical studies and are linked to the release into the circulatory system of compounds (releasable) located at the interface MD / biological medium or MD/drug formulation. Understanding and anticipating these risks requires an in-depth knowledge of the materials used in the manufacture of MDs. It is thus entirely accepted that the success of an assessment of the biological risks of an MD is directly linked to that of its physicochemical characterisation, implying these surface, volume properties and its composition. Therefore, such an approach necessarily requires the collaboration of specialists in the physicochemical of polymers, analytical chemists and biologists all with a common core of skills: knowledge of the MD.
Share the article

Katia MANERLAX, Lionel TORTOLANO & Najet YAGOUBI – Univ.Paris Saclay, UFR de Pharmacie, Lab. Matériaux & Santé EA 401.
References
Exudation of additives to the surface of medical devices: impact on biocompatibility in the case of polyurethane used in implantable catheters. J Biomed Mater Res A., Volume104 (12), 2016, p. 2954- 2967
Interaction of intraocular lenses with fibronectin and human lens epithelial cells: Effect of chemical composition and ageing. J Biomed Mater Res A., volume 103(12), 2015, p.3843-51
