


Summary
- Injectable Combination Products. Issues and challenges for industry.
- Deadline Extension or Not: Key Points for Class I Manufacturers
- The new challenges of injectable medicines
- Clinical Investigation of Combined Medical Devices Under New Regulation (EU) 2017/745 (MDR)
- Contribution of the physicochemical characterization of the materials constituting medical devices for rationalisation of their biological assessment.
- Steering Cleaning Validation Performance : A Key Industrial Challenge.
- Cleaning Process Validation: Why and How to Validate Analytical Methods and Related Sampling Methods
- A Risk-Based Approach to Stainless Steel Equipment Maintenance in cGMP Manufacturing Environment
- Single Use Systems vs Re-Usable Stainless-Steel Equipment. Compliance & Quality Perspective.
Clinical Investigation of Combined Medical Devices Under New Regulation (EU) 2017/745 (MDR).

1. CE Marking
What is a combined medical device?
A combined medical device is defined by the European regulations as being a device incorporating, as an integral part, a substance which, if used separately, can be considered to be a medicinal product,(1),including a medicinal product derived from human blood or human plasma, as defined in Point 10 of Article 1 of Directive 2001/83/CE, and the action of which is ancillary to that of the device. In this case, the manufacturer must demonstrate that the principal mode of action is that of the device (a “mechanical” type of action), with the medicinal substance having only an ancillary role (action of a pharmacological, immunological or metabolic type).
Some examples of combined devices:
• A coronary stent comprising a coating containing heparin: the principal mode of action is that of the stent which, by its mechanical (“spring”) action, dilates the coronary arteries to restore blood flow. The heparin (medicinal substance) has a secondary or ancillary action aiming to reduce the risk of a blood clot forming when the stent is inserted;
• A bone substitute (hydroxyapatite) containing an antibiotic (gentamicin): the main action is that of the bone substitute which serves to fill in any missing bone during an orthopedic operation. The gentamicin (medicinal substance) has an ancillary action, the purpose of which is to reduce the risk of bone infection during an operation that might favor such a risk (on an open fracture, for example).
Medical devices (MD) are classified in one of the following four classes according to their level of risks to the patient and user: I (the lowest-risk MDs), IIa, IIb and III (the highest-risk MDs).Combined MDs are in Class III by virtue of Rule 14 in Annex VIII to Regulation (UE) 2017/745: “Alldevices incorporating, as an integral part, a substance which, if used separately, can be considered to be a medicinal product, as defined in point 2 of Article 1 of Directive 2001/83/EC, including a medicinal product derived from human blood or human plasma, as defined in point 10 of Article 1 of that Directive, and that has an action ancillary to that of the devices, are classified as class III.”
CE marking of combined medical devices
Like all medical devices, a combined device must obtain a CE marking to allow it to be marketed in Europe and in those third countries that recognize the CE marking (Switzerland, Turkey, Norway, etc.). The CE mark attests to the performances and conformity to the General Safety and Performance Requirements (GSPR -Annex I to the MDR) of the device, as regards the safety and health of patients or third-party users. It is the responsibility of the manufacturer to demonstrate this conformity. In addition, as a combined device is a Class III device, an assessment by a third-party body, known as a Notified Body (NB) and appointed by the competent authority in the country in which the NB is based, is necessary before the CE label can be affixed.
Initial submission for the CE mark
Within the framework of the conformity assessment procedure for an initial CE mark application for a Class III combined medical device, the manufacturer must submit a Technical File (TF) for the device to the notified body. Compared to the TF for a “non-combined” device, this one is different in that it comprises a specific section on the medicinal substance which is divided into five parts:
- Administrative information;
- Summaries of the file comprising the risk analysis, the rationale for the presence of the medicinal substance, the description of the modes of action of the components of the medical device, distinguishing between those linked to the device, to the medicinal substance and to the combination of the two; expert reports (quality, non-clinical and clinical);
- The quality part concerning the medicinal substance alone and the medicinal substance incorporated into the device;
- The pre-clinical part;
- The clinical part.
The manufacturer must provide evidence of the usefulness of incorporating the medicinal substance into the medical device. The notified body then checks this usefulness.
After checking the usefulness of the substance as a part of the device and taking account of the intended purpose of the device, the notified body asks one of the national competent authorities designated by the Member States or the EMA (European Medicines Agency) for a scientific opinion on the quality and safety of the medicinal substance, including on the benefit and/or risk linked with the incorporation of the substance into the medical device. These competent authorities or the EMA are referred to as the “medicinal products authority consulted”.
For those medical devices incorporating a medicinal substance that may be considered to be a medicinal product subject to a centralized market authorization procedure or a medicinal product derived from human blood or plasma, the EMA will be the medicinal products authority consulted.
For medical devices incorporating another medicinal substance, the medicinal products authority consulted may be either a competent national authority or the EMA. The medicinal products authority consulted then has 210 days from receipt of all the necessary documentation to assess the file and provide its scientific opinion, taking account of the manufacturing process and the data on the usefulness of incorporating the substance into the medical device, as determined by the notified body. The scientific opinion of the medicinal products authority consulted, and any update of that opinion, shall be included in the documentation of the notified body concerning the device. The notified body shall give due consideration to the views expressed in the scientific opinion when making its decision. The notified body shall not deliver the certificate if the scientific opinion is unfavorable and shall convey its final decision to the medicinal products authority consulted.
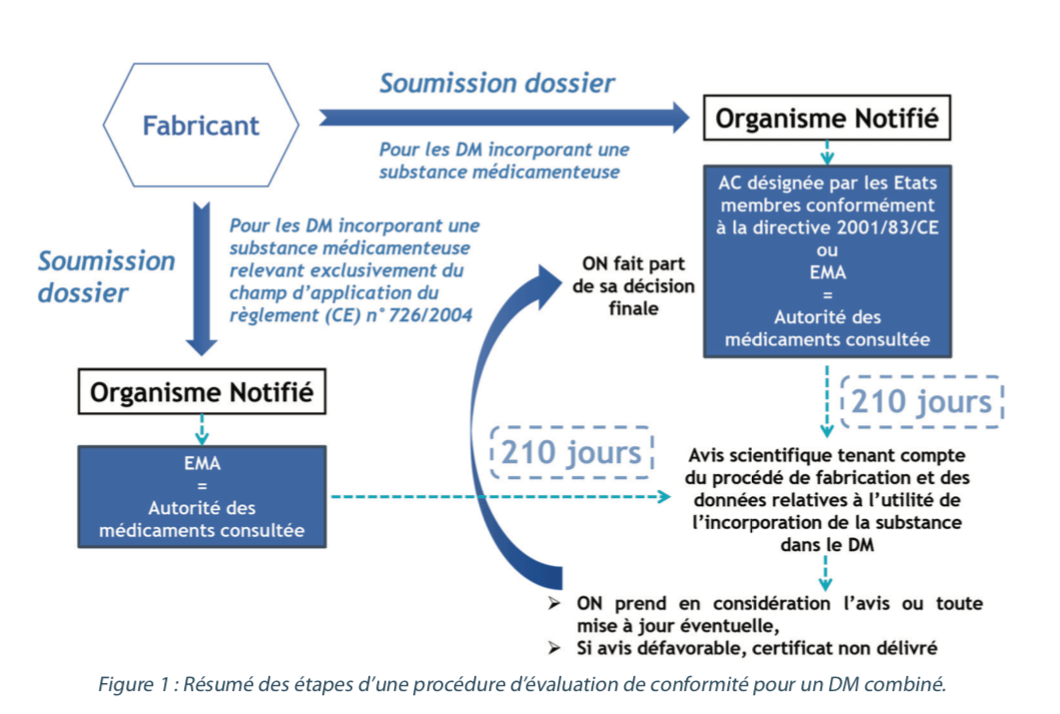
Management of post-CE marking modifications concerning the active substance
a) At the initiative of the combined MD manufacturer
Once the CE marking has been obtained, if the manufacturer should decide to make any modifications affecting the medicinal substance incorporated into the medical device, such as to its manufacturing process, it must inform the notified body. The notified body shall seek the opinion of the medicinal products authority consulted to confirm that the quality and safety of the substance remain unchanged. in order to confirm that the quality and safety of the ancillary substance remain unchanged. That authority shall provide its opinion within 60 days. The medicinal products authority consulted shall assess whether the changes have any negative impact on the risk and/or benefit previously established concerning the incorporation of the substance into the device. If the scientific opinion is unfavorable, the notified body shall not deliver the supplement to the technical documentation assessment certificate.
b) At the initiative of the competent authority
If the medicinal products authority consulted obtains information on the ancillary medicinal substance, which could have an impact on the risk and/or benefit previously established concerning the incorporation of the substance into the device, it shall advise the notified body as to whether this information has an impact on the risk and/or benefit previously established concerning the incorporation of the substance into the device. The notified body shall take that advice into account and reconsider its conformity assessment.
2. Clinical investigation into a combined medical device
Regulatory background
For an initial CE marking procedure, the clinical investigation (clinical trial) will be mandatory for all Class III devices, except in the following cases:
- If the device was designed by modifying a device already marketed by the same manufacturer and the manufacturer has demonstrated that the modified device is equivalent to the marketed device, provided that the clinical evaluation of the marketed device is sufficient to demonstrate conformity of the modified device with the General Safety and Performance Requirements (GSPR);
- A manufacturer of a device demonstrated to be equivalent to an already marketed device not manufactured by him is not required to perform a clinical investigation, provided that the following conditions are fulfilled: the two manufacturers have signed a contract that explicitly allows the manufacturer of the second device full access to the technical documentation, and the original clinical evaluation was performed in compliance with the requirements of the MDR.
For MDs that already have the CE mark in accordance with Directive 93/42/EEC, the requirement to perform clinical investigations does not apply to class III devices and implantable devices for which the clinical evaluation is:
- based on sufficient clinical data, and
- in compliance with the relevant product-specific common specification for the clinical evaluation of that kind of device, where such a specification is available.
Given the requirements described above, the clinical investigation is mandatory in the majority of cases for a CE mark application for a combined medical device (Class III).
For all Class III devices, the manufacturer may, before conducting the clinical evaluation and/or investigation, consult an expert panel with the aim of reviewing the manufacturer’s intended clinical development strategy and proposals for clinical investigation.(2) In order to secure the clinical development strategy, it would seem appropriate that this opinion should be collected before launching the clinical investigation.
In order to establish the conformity of a Class III combined medical device with the GSPR in the MDR, the manufacturer must carry out clinical investigations in compliance with the ethical principles of the Member States. The clinical investigation serves to establish and verify that, under normal conditions of use, a device achieves the performance as specified by its manufacturer. It also serves to establish and verify both the benefits and risks of a device, and the clinical safety of the device. The clinical investigation also serves to detect any undesirable side-effects of the device in normal conditions of use and to assess whether they constitute acceptable risks when weighed against the expected benefits.
Clinical investigation
The documents to be provided to obtain an authorization for a clinical 41 investigation (3)
I. Application form
This form contains, among other information, the name, address and contact details of the sponsor and, where they are different, those of the manufacturer of the device intended for clinical investigation. It also contains the title of the clinical investigation and a summary of the clinical investigation plan (for a first application), the details and/or references of the clinical investigation plan and a brief description of the device and its classification.
The sponsor must mention the Member States and third countries in which the investigation is to be conducted, as part of a multicenter or multinational study.
The sponsor shall provide a brief description of the investigational device, its classification and other information necessary for the identification of the device and device type.
If the notified body is already involved at the stage of the clinical investigation application, the sponsor must provide details to identify it.
II. Investigator’s brochure
The Investigator’s Brochure shall contain the clinical and non-clinical information on the investigational device that is relevant for the clinical investigation and available at the time of application for the authorization.
It must be clearly identified and contain in particular the following information:
- Data to identify and describe the device, including information on its intended purpose, classification, design and manufacture;
- The manufacturer’s instructions for installation, maintenance, maintaining hygiene standards (storage and handling requirements), as well as the available information to be placed on the label and instructions;
- A pre-clinical evaluation based on relevant pre-clinical testing data (notably biocompatibility, performance tests, sterilization validation, stability data, etc.) and experimental data;
- Existing clinical data, in particular from relevant scientific literature relating to the performance, clinical benefits to patients, design characteristics and intended purpose of the device , and all other relevant data;
- A summary of the benefit-risk analysis and the risk management, including information on known or foreseeable risks, any undesirable effects, contraindications and warnings;
- For combined medical devices incorporating a medicinal substance, detailed information on the medicinal substance and compliance with the GSPR, and on the specific risk management in relation to the substance, as well as evidence for the added value of incorporation of such substance in relation to the clinical benefit and/or safety of the device;
- A list detailing the GSPR, including the standards and common specifications applied, as well as a description of the solutions for fulfilling the GSPR when those standards or common specifications have not been fulfilled;
- A detailed description of the clinical procedures and diagnostic tests used in the course of the clinical investigation, and in particular information on any deviation from normal clinical practice.
III. The clinical investigation plan
The Clinical Investigation Plan sets out the rationale, objectives, design, methodology, monitoring and conduct of the clinical investigation, as well as record-keeping and the method of analysis for it.
In this document, the sponsor must indicate the single identification number of the clinical investigation, the identification of the sponsor and of the principal investigator at each investigational site. It describes how the clinical investigation is financed and the overall synopsis of the clinical investigation.
The plan sets out information identifying and describing the device, including its intended purpose, its manufacturer, its traceability, the target population, materials coming into contact with the human body, a review of the background literature, the current state of the art in clinical care in the relevant field of application and the proposed benefits of the new device.
The sponsor must provide information on the objectives and hypotheses of the clinical investigation, the design of the clinical investigation and general information such as type of investigation with the rationale for choosing it, its endpoints and its variables. It must also provide information on subjects, selection criteria, the size of investigation population and its representativeness in relation to the target population.
The Clinical Investigation Plan also contains a presentation of the different tests for statistical analysis.
It also presents a monitoring plan and a data management plan, a policy regarding follow-up and management of any deviations from the investigation plan at the investigational site and clear prohibition of use of waivers. It also contains a statement of compliance with the recognized ethical principles for medical research involving humans, and the principles of good clinical practice in the field of clinical investigations of devices, as well as with the applicable regulatory requirements. The informed consent of the patient must be presented.
The sponsor must describe the criteria and procedures for follow-up of subjects following the end, a temporary halt or the early termination of an investigation, for follow-up of subjects who have withdrawn their consent and procedures for subjects lost to follow-up. A description must also be provided of the arrangements for taking care of the subjects after their participation in the clinical investigation has ended, where additional care is necessary because of the subjects’ participation in the clinical investigation and where it differs from that normally expected for the medical condition in question.
Other information must also be included in the clinical investigation plan, such as the signed statement by the natural or legal person responsible for the manufacture of the device, the documents to be used to obtain informed consent, including the patient information sheet and the informed consent document.
IV. Timelines for appraisal of authorization applications
The sponsor of the clinical investigation shall submit an application to each Member State in which the clinical investigation is to be conducted, accompanied by the documentation referred to above. The Member State concerned shall have ten days to assess acceptability and the application that has been submitted. If it deems that the clinical investigation does not fall within the scope of the MDR or if the dossier is incomplete, it shall inform the sponsor to this effect. The sponsor then has ten days to comment or to complete its application. The Member State concerned may extend this period by a maximum of 20 days.
If the sponsor has not provided comments nor completed the application within the time limit, the application will be cancelled.
If the sponsor considers the application does fall under the scope of the MDR and/or is complete but the Member State concerned is not of the same opinion, the application shall be considered to have been rejected. The Member State concerned shall provide for an appeal procedure in respect of such refusal.
Within five days of receiving the comments or requested additional information, the Member State concerned shall notify the sponsor whether the clinical investigation is considered as falling within the scope of this Regulation and the application is complete. The date on which the sponsor is notified corresponds to the validation date of the application. If the sponsor is not notified, the validation date shall be the last day of the period set by the Member State concerned.
The sponsor may start the clinical investigation as soon as the Member State concerned has notified the sponsor of its authorization, and provided that the ethics committee in the Member State concerned has not issued a negative opinion in respect of the clinical investigation. The Member State shall notify the sponsor of the authorization within 45 days of the validation date.(4)
It should be noted that the MDR also makes provision for the possibility of a coordinated assessment procedure to obtain authorization to conduct clinical investigations (5).
V. The clinical investigation report
At the end of the clinical investigation, the sponsor must provide a report containing the results of the investigation. The clinical investigation report is the most important document in the evaluation of the safety and clinical performances of the medical device, as claimed by the manufacturer.
Conclusion
The application of the MDR in May 2020 is going to tighten the rules on the clinical evaluation of medical devices, and more particularly those in Class IIb implantable and Class III (including combined MDs).
For a combined device in Class III, it will no longer be possible, except in very rare cases, to base a clinical evaluation on a critical review of publications on the safety and performances of a device that is equivalent to the device undergoing clinical evaluation (provided that equivalence with the evaluated device has been demonstrated).
The conduct of such a clinical investigation will therefore be mandatory to obtain the CE mark and market the combination medical device.
Share article


Michel HUC – ASPE Conseil
michel.huc@aspe-conseil.eu
Glossary
CA: Competent Authority
CE: European Conformity
CEE: European Economic Community
EMA : European Medicines Agency
EU: European Union
GSPR: General Safety and Performance Requirements
MD: Medical Device
MDR: Medical Devices Regulation [(EU) 2917/745]
NB: Notified Body
TF: Technical File
References
1. Within the meaning of point 2 of Article 1 of Directive 2001/83/CE
2. Chapter VI Article 61 of Regulation (EU) 2017/745
3. Annex XV Chapter II of Regulation (EU) 2017/745
4. Chapter VI Article 70 of Regulation (EU) 2017/745
5. Chapter VI Article 78 of Regulation (EU) 2017/745
