


Sommaire
- Produits Combinés Injectables. Enjeux et challenges pour les industriels.
- Extension des délais ou non, ce que les fabricants de Classe I doivent retenir.
- Les nouveaux défis des médicaments injectables.
- L’investigation clinique des dispositifs médicaux combinés dans le cadre du nouveau règlement (UE) 2017/745 (RDM).
- Apport de la caractérisation physico-chimique des matériaux constitutifs des dispositifs médicaux pour une rationalisation de leur évaluation biologique.
- Piloter la performance des validations de nettoyage : un enjeu industriel fort.
- Validation des procédés de nettoyage : pourquoi et comment valider les méthodes analytiques et de prélèvements associées.
- Maintenance des équipements en acier inoxydable dans un environnement de fabrication conforme aux BPF : une approche basée sur les risques.
- Single Use Systems vs Re-Usable Stainless-Steel Equipment. Compliance & Quality Perspective.
L’investigation clinique des dispositifs médicaux combinés dans le cadre du nouveau règlement (UE) 2017/745 (RDM).

1. Le marquage CE
Qu’est-ce qu’un dispositif médical combiné ?
Un dispositif médical combiné est défini par la règlementation Européenne comme étant un dispositif incorporant comme partie intégrante une substance qui, utilisée séparément, peut être considérée comme un médicament(1), y compris un médicament dérivé du sang ou du plasma humain tel que défini à l’article 1er, point 10, de la directive 2001/83/CE, et dont l’action est accessoire à celle du dispositif. Dans ce cas, le fabricant doit démontrer que l’action principale est celle du dispositif (avec un mode d’action de type “mécanique”), la substance médicamenteuse n’ayant qu’un rôle accessoire (action de type pharmacologique, immunologique ou métabolique).
Quelques exemples de dispositifs médicaux combinés :
• Stent coronarien comportant un revêtement contenant de l’héparine : l’action principale est celle du stent qui grâce à son action mécanique (“ressort”) dilate les artères coronaires permettant de rétablir la circulation sanguine. L’héparine (substance médicamenteuse) a une action secondaire ou accessoire, visant à diminuer le risque de thrombus lors de la pose du stent ;
• Substitut osseux (hydroxyapatite) contenant un antibiotique (gentamicine) : l’action principale est celle du substitut osseux qui sert à combler les manques osseux lors d’une intervention d’orthopédie. La gentamicine (substance médicamenteuse) a une action accessoire dont le but est de diminuer le risque d’infection osseuse lors d’une intervention qui peut favoriser ce type de risque (sur une fracture ouverte par exemple).
Les dispositifs médicaux (DM) sont classés selon leur niveau de risques pour le patient et l’utilisateur, dans une des quatre classes suivantes : I (DM les moins à risques), IIa, IIb et III (DM les plus à risques). Les DM combinés sont de classe III en vertu de la règle 14 de l’annexe VIII du règlement (UE) 2017/745 : “Tous les dispositifs incorporant comme partie intégrante une substance qui, utilisée séparément, peut être considérée comme un médicament au sens de l’article 1er, point 2, de la directive 2001/83/CE, y compris un médicament dérivé du sang ou du plasma humain tel que défini à l’article 1er, point 10, de ladite directive, et dont l’action est accessoire à celle des dispositifs, relèvent de la classe III”
Le marquage CE des dispositifs médicaux combinés
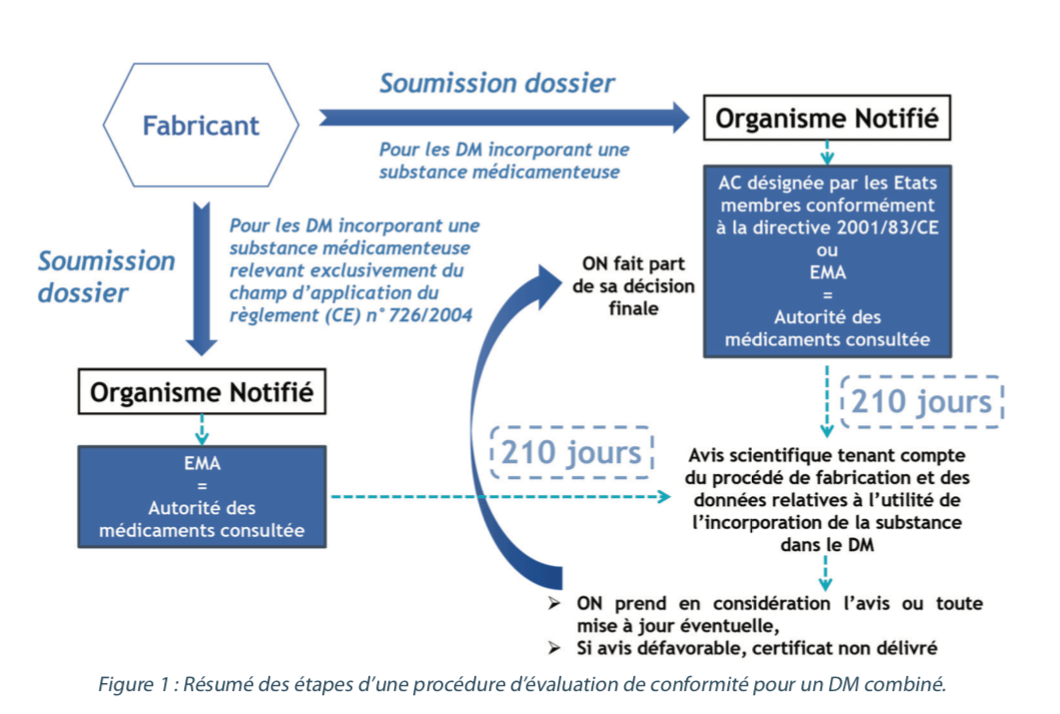
Comme pour tous les dispositifs médicaux, un dispositif médical combiné doit obtenir un marquage CE afin de pouvoir être commercialisé en Europe, et dans les pays tiers reconnaissant le marquage CE (Suisse, Turquie, Norvège, …). Le marquage CE permet d’attester des performances et de la conformité aux exigences générales en matière de sécurité et de performance (EGSP – Annexe I du RDM) du dispositif, en ce qui concerne la sécurité et la santé des patients ou des tiers utilisateurs. La démonstration de cette conformité est de la responsabilité du fabricant. De plus, comme un dispositif médical combiné est un dispositif de classe III, une évaluation par un organisme tiers, appelé organisme notifié (ON), désigné par l’autorité compétente du pays dans lequel l’ON est basé, est nécessaire avant de pouvoir apposer le marquage CE.
La demande initiale de marquage CE
Dans le cadre de la procédure d’évaluation de la conformité, lors d’une demande initiale de marquage CE pour un dispositif médical combiné de classe III, le fabricant doit soumettre le dossier technique (DT) du dispositif à un organisme notifié. Ce dossier technique présente la particularité, par rapport au DT d’un DM “non combiné”, de comporter une partie spécifique à la substance médicamenteuse qui est divisée en cinq parties :
• Les informations administratives ;
• Les résumés du dossier comportant l’analyse de risque, la justification de la présence de la substance médicamenteuse, la description des modes d’action des composants du dispositif médical en distinguant ceux liés au dispositif, à la substance médicamenteuse et à la combinaison des deux ; les rapports d’experts (qualité, non-clinique et clinique) ;
• La partie qualité concernant la substance médicamenteuse seule et la substance médicamenteuse incorporée dans le dispositif ;
Après avoir vérifié l’utilité de la substance en tant que partie du dispositif, et en tenant compte de la destination du dispositif, l’organisme notifié demande à l’une des autorités compétentes nationales désignées par les États membres ou à l’EMA (European Medicines Agency) un avis scientifique sur la qualité et la sécurité de la substance médicamenteuse, y compris sur le bénéfice et/ou le risque liés à l’incorporation de cette substance dans le dispositif médical. Les autorités compétentes ou l’EMA sont dénommées “autorité des médicaments consultée”.
Pour les dispositifs médicaux incorporant une substance médicamenteuse susceptible d’être considérée comme un composant de médicament relevant d’une procédure d’autorisation de mise sur le marché centralisée ou un médicament dérivé du sang ou du plasma humain, l’EMA sera l’autorité des médicaments consultée.
Pour les dispositifs médicaux incorporant une substance médicamenteuse autre, l’autorité des médicaments consultée pourra être soit une autorité compétente nationale, soit l’EMA. L’autorité des médicaments consultée dispose ensuite de 210 jours, à compter de la réception de l’ensemble de la documentation nécessaire, pour évaluer le dossier et rendre un avis scientifique en tenant compte du procédé de fabrication et des données relatives à l’utilité de l’incorporation de la substance dans le dispositif médical telle qu’elle a été déterminée par l’organisme notifié. L’organisme notifié inclut dans sa documentation, relative au dispositif médical combiné, l’avis scientifique de l’autorité des médicaments consultée, ainsi que toute mise à jour éventuelles de cet avis. Lorsque l’organisme notifié rend sa décision, il prend en considération les points exprimés dans l’avis scientifique. Si l’avis scientifique est défavorable, l’organisme notifié ne délivrera pas le certificat CE. Sa décision finale sera transmise à l’autorité des médicaments consultée.
Gestion des modifications post marquage CE, touchant la substance active
a) A l’initiative du fabricant du DM combiné
Après obtention du marquage CE, si le fabricant décide d’apporter des modifications affectant la substance médicamenteuse incorporée dans un dispositif médical, comme, par exemple, son procédé de fabrication, il doit en informer l’organisme notifié. Ce dernier demande l’avis de l’autorité des médicaments consultée, qui rend son avis dans les 60 jours, afin d’obtenir la confirmation que la qualité et la sécurité de la substance restent inchangées. L’autorité des médicaments consultée évalue si les modifications ont des incidences négatives sur le risque et/ou le bénéfice précédemment établis concernant l’incorporation de la substance dans le dispositif. Si l’avis scientifique est défavorable, l’organisme notifié ne délivre pas le document complémentaire au certificat d’évaluation de la documentation technique.
b) A l’initiative de l’autorité compétente
Si l’autorité des médicaments consultée obtient des informations sur la substance médicamenteuse accessoire susceptibles d’avoir une incidence sur le risque et/ou le bénéfice précédemment établis, elle donne un avis à l’organisme notifié sur le fait de savoir si les informations ont une incidence sur le risque et/ou le bénéfice précédemment établis concernant l’incorporation de la substance dans le dispositif. L’organisme notifié prend en compte cet avis et reconsidère son évaluation de la conformité.
2. L’investigation clinique d’un dispositif médical combiné
Contexte réglementaire
Dans le cadre d’une procédure initiale de marquage CE, l’investigation clinique (essai clinique) sera obligatoire pour tous les dispositifs de classe III, sauf dans les cas suivants :
• Si le dispositif a été conçu en modifiant un dispositif déjà commercialisé par le même fabricant et que le fabricant a démontré que le dispositif modifié est équivalent au dispositif déjà commercialisé et que l’évaluation clinique du dispositif commercialisé suffit à démontrer la conformité du dispositif modifié avec les Exigences Générales en matière de Sécurité et de Performances (EGSP) ;
• Le fabricant d’un dispositif pour lequel il a été démontré qu’il est équivalent à un dispositif déjà commercialisé et non fabriqué par lui, n’est pas dans l’obligation de conduire d’investigation clinique, pour autant que les conditions suivantes soient remplies : les deux fabricants ont conclu un contrat qui accorde explicitement au fabricant du second DM un accès total et permanent à la documentation technique, et l’évaluation clinique d’origine a été effectuée conformément aux exigences du RDM.
Pour les DM déjà marqués CE, selon la directive 93/42/CEE, l’obligation de conduire des investigations cliniques ne s’applique pas aux dispositifs de classe III et aux dispositifs implantables pour lesquels l’évaluation clinique :
• est fondée sur des données cliniques suffisantes, et
• est conforme à la spécification commune par produit qui est applicable pour l’évaluation clinique de ce type de dispositif, lorsqu’il en existe une.
Compte tenu des exigences décrites plus haut, l’investigation clinique est obligatoire dans la majeure partie des cas, dans le cadre d’une demande de marquage CE, pour un dispositif médical combiné (classe III).
Pour tous les dispositifs de classe III, le fabricant peut, avant d’effectuer son évaluation clinique et/ou son investigation clinique consulter un groupe d’experts dans le but d’examiner la stratégie de développement clinique prévue par le fabricant et les propositions d’investigation clinique (2). Afin de sécuriser la stratégie de développement clinique, il semble pertinent de recueillir cet avis, préalablement au lancement de l’investigation clinique.
Afin d’établir la conformité aux EGSP du RDM d’un dispositif médical combiné de classe III, le fabricant doit réaliser des investigations cliniques tout en respectant les principes éthiques des États membres. L’investigation clinique permet d’établir et de vérifier que, dans des conditions normales d’utilisation, le dispositif atteint les performances prévues telles qu’elles sont spécifiées par son fabricant. Elle permet également d’établir et de vérifier, à la fois les bénéfices et les risques d’un dispositif et la sécurité clinique de ce même dispositif. L’investigation clinique permet aussi de détecter les éventuels effets secondaires indésirables du dispositif dans des conditions normales d’utilisation et d’évaluer si ces derniers constituent un risque acceptable comparé aux bénéfices attendus.
L’investigation clinique
Les documents à fournir pour l’obtention d’une autorisation d’investigation clinique (3)
I. Formulaire de demande d’autorisation
Ce formulaire contient notamment le nom, l’adresse et les coordonnées du promoteur ; et, s’ils sont différents, ceux du fabricant du dispositif médical faisant l’objet de l’investigation clinique. Il contient également l’intitulé de l’investigation clinique ainsi qu’un résumé du protocole d’investigation, le statut de la demande d’investigation clinique (dans notre cas première demande), les détails et/ou les références du plan d’investigation clinique, une description succincte du dispositif ainsi que sa classification.
Le promoteur doit mentionner les Etats membres et les pays tiers dans lesquels l’investigation clinique doit être menée, dans le cadre d’une étude multicentrique ou multinationale.
Le promoteur fournira une description succincte du dispositif faisant l’objet de l’investigation clinique, sa classification et d’autres informations nécessaires aux fins de l’identification et du type de dispositif.
Si l’organisme notifié est déjà impliqué au stade de la demande d’investigation clinique, le promoteur doit fournir des informations permettant de l’identifier.
II. Brochure pour l’investigateur
La brochure pour l’investigateur contient les données non cliniques et cliniques concernant le dispositif médical qui sont utiles à l’investigation clinique et disponibles au moment de la demande d’autorisation.
Elle doit être clairement identifiée et doit contenir notamment :
• Les données d’identification et la description du dispositif, avec des informations sur sa destination, sa classification, sa conception et sa fabrication ;
• Les instructions du fabricant concernant l’installation, la maintenance, le maintien des normes d’hygiène et l’utilisation (condition de stockage et de manipulation) ainsi que les données disponibles devant figurer sur l’étiquette et la notice ;
• Une évaluation préclinique fondée sur les données des essais précliniques (notamment la biocompatibilité, les essais de performance, la validation de la stérilisation, les données de stabilité …) et les données expérimentales pertinentes ;
• Les données cliniques existantes, provenant notamment de la littérature scientifique pertinente relative aux performances, aux bénéfices cliniques pour les patients, aux caractéristiques de conception et à la destination du dispositif ; ainsi que toutes les autres données pertinentes ;
• Un résumé de l’analyse bénéfice/risque et de la gestion des risques, et notamment des informations sur les risques connus ou prévisibles, les effets indésirables, les contre-indications et les mises en garde ;
• Pour les dispositifs médicaux combinés, incorporant une substance médicamenteuse, des informations détaillées sur la substance médicamenteuse, ainsi que sur le respect des EGSP et sur la gestion des risques particuliers posés par la substance, et également des éléments de preuve concernant la valeur ajoutée que présente l’incorporation de cette substance en termes de bénéfice et/ou de sécurité du dispositif ;
• Une liste détaillant les EGSP, y compris les normes et les spécifications communes appliquées, ainsi qu’une description des solutions retenues pour satisfaire aux EGSP lorsque les normes ou les spécifications communes n’ont pas été respectées ;
• Une description détaillée des procédures cliniques et des essais diagnostiques utilisés au cours de l’investigation clinique, et notamment, des informations de tout écart par rapport à la pratique clinique courante.
III. Le protocole d’investigation clinique
Le protocole d’investigation clinique présente la justification, les objectifs, la conception, les méthodologies, le contrôle, et les modalités de conduite de l’investigation clinique, ainsi que la documentation de ses résultats et la méthode d’analyse la concernant.
Dans ce document, le promoteur doit renseigner le numéro d’investigation unique, les données d’identification du promoteur et de l’investigateur principal sur chaque site d’investigation. Il décrit les modalités de financement de l’investigation clinique, ainsi que le scénario général de l’investigation clinique.
Dans ce protocole, sont renseignées les données d’identification et la description dudispositif, notamment sa destination, son fabricant, sa traçabilité, la population cible, les matériaux entrant en contact avec le corps humain, l’examen de la documentation générale, l’état de l’art concernant les soins cliniques dans le domaine d’application concerné et les bénéfices escomptés du nouveau dispositif.
Le promoteur doit renseigner les objectifs et les hypothèses de l’investigation clinique, la conception de l’investigation clinique et les informations générales, comme le type d’investigation et les raisons justifiant ce choix, les critères de son évaluation et ses variables. Il doit également renseigner les informations sur les participants, les critères de sélection, la taille de la population visée par l’investigation et sa représentativité par rapport à la population cible.
Le protocole d’investigation contient aussi une présentation des différents tests permettant l’analyse statistique.
Le protocole expose un plan de surveillance et un plan de gestion des données, une politique en matière de suivi et de gestion de tout écart par rapport au protocole d’investigation clinique sur le site d’investigation et une interdiction claire du recours à toute dérogation.
Le protocole contient également la déclaration de conformité avec les principes éthiques reconnus applicables à la recherche médicale impliquant les êtres humains et avec les principes des bonnes pratiques en matière d’investigations cliniques des dispositifs, ainsi qu’avec les exigences de la réglementation applicables. Le consentement éclairé du patient doit être présenté.
Le promoteur doit décrire les critères et les procédures de suivi des participants après la fin, l’interruption temporaire ou l’arrêt anticipé de l’investigation clinique, de suivi des participants qui ont retiré leur consentement, ainsi que les procédures concernant les participants qui échappent au suivi. Doivent être également décrites les modalités de prise en charge des participants une fois leur participation à l’investigation clinique terminée, lorsque des soins supplémentaires sont nécessaires du fait de leur participation à l’investigation clinique, dans le cas où ces soins diffèrent des soins attendus par rapport à l’affection concernée.
D’autres informations doivent figurer dans le protocole d’investigation clinique comme la déclaration signée par la personne physique ou morale responsable de la fabrication du dispositif, les documents à utiliser aux fins de l’obtention d’un consentement éclairé, y compris la fiche d’information du patient et le document relatif au consentement éclairé.
IV. Les délais d’instruction des demandes d’autorisation
Le promoteur de l’investigation clinique introduit, auprès de chaque État membre concerné par l’investigation, une demande accompagnée de tous les documents précédemment cités. L’État membre concerné dispose de dix jours pour évaluer la recevabilité et la demande introduite. S’il estime que l’investigation clinique n’est pas en accord avec le RDM ou si le dossier est incomplet, il en informe le promoteur.
Le promoteur a alors de dix jours pour formuler des observations ou compléter sa demande. L’État membre concerné peut prolonger ce délai d’une vingtaine de jours au maximum.
Si le promoteur ne formule pas d’observations ni ne complète la demande dans le délai, la demande est annulée.
Si le promoteur estime que sa demande relève du RDM et/ou qu’elle est complète mais que l’État membre concerné n’est pas du même avis, la demande est rejetée. L’État membre concerné prévoit alors une procédure de recours pour un tel refus.
Dans délai de cinq jours suivant la réception des observations ou des informations complémentaires demandées, l’État membre concerné indique au promoteur si l’investigation clinique est réputée relever du RDM et si la demande est complète. La date à laquelle le promoteur est informé correspond à la date de validation de la demande. Si le promoteur n’est pas informé, la date de validation correspond au dernier jour du délai fixé par l’État membre concerné.
Le promoteur peut débuter l’investigation clinique dès que l’État membre concerné a notifié son autorisation au promoteur et pour autant que le comité d’éthique dans l’État membre concerné n’ait pas émis d’avis défavorable concernant l’investigation clinique. L’État membre concerné notifie l’autorisation au promoteur dans un délai de quarante-cinq jours suivant la date de validation (4).
Il faut noter que le RDM prévoit également la possibilité d’avoir recours à une procédure d’évaluation coordonnée pour obtenir l’autorisation de réaliser les investigations cliniques (5).
V. Le rapport d’investigation clinique
A la fin de l’investigation clinique, le promoteur doit fournir un rapport contenant les résultats de cette investigation clinique. Le rapport d’investigation clinique constitue le document le plus important dans l’évaluation de la sécurité et des performances cliniques du dispositif médical, telles que revendiquées par le fabricant.
Conclusion
L’application du RDM, en mai 2020, va durcir les règles de l’évaluation clinique des dispositifs médicaux, et plus particulièrement ceux des classes IIb implantables et III (dont les DM combinés).
En effet, fonder une évaluation clinique sur un examen critique des publications relatives à la sécurité et aux performances d’un dispositif équivalent au dispositif objet de l’évaluation clinique (à condition que l’équivalence avec le dispositif évalué soit démontrée), pour un dispositif médical combiné de classe III, ne sera plus possible, sauf dans de très rares cas.
La réalisation d’une investigation clinique sera donc obligatoire pour pouvoir obtenir le marquage CE, et commercialiser le DM combiné.
Partager l’article


Michel HUC – ASPE Conseil
Michel Huc, Docteur en Pharmacie et titulaire d’un MBA, a occupé de multiples responsabilités dans l’industrie pharmaceutique (responsable contrôle qualité, directeur de site, directeur des opérations industrielles, pharmacien responsable d’un laboratoire pharmaceutique en charge des divisions R&D, Affaires Réglementaires et Assurance Qualité) et celle des dispositifs médicaux.
En 2007, il fonde Aspe Conseil, cabinet de conseil qui propose des services en R&D, Affaires Réglementaires et Qualité à ses clients dans le domaine des produits de santé (www.aspe-conseil.eu) afin de leur permettre de répondre aux exigences réglementaires et normatives à chaque étape du cycle de vie de leurs produits.
michel.huc@aspe-conseil.eu
Glossary
References
1978 (US CFR): Section 211.67 added describing equipment cleaning and maintenance
1993 (US FDA guideline): Guide to inspections for cleaning validation2000 (ICH Q7): Guide for active pharmaceutical ingredients
2004 (US FDA guide): Guide to inspections validation of cleaning processes
2010 (PDA): TR 49 – Points to consider for Biotechnology Cleaning Validation
2010 (ISPE): Risk-Based Manufacture of Pharmaceutical Products
2012 (PDA): TR 29 – Points to consider for Cleaning Validation
2015 (EMA): Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities
2015 (Eudralex Vol. 4 – GMP Guidelines Annex 15): Qualification and Validation
