


Summary
- Remote Audit. Overview of communications from health authorities on remote inspections and audits
- Hesitant beginnings with a sustainable method. Experience feedback
- Remote Audit. The role of the Covid crisis in its generalization, principles, technologies and auditor profiles
- The remote audit: strengths & weaknesses
- Microbiological study on the management of holes in gloves for isolators
- Contamination Control Strategy: practices & a case study of a CCS implementation
- Toxicological approach to define the PDE for your cleaning validation process
- How high performance materials for secondary packaging components can significantly reduce particle contamination in Ready-to-use vials
Contamination Control Strategy: practices & a case study of a CCS implementation.
As a result of the successive revisions to the draft EU GMP Annex 1(1), the term “Contamination Control Strategy (CCS)” has become as well known as the term “Quality Risk Management (QRM),” which is intriguing because the CCS foundation is built on QRM principles.

Since the release of the first EU GMP Annex 1 draft, many questions have been raised by manufacturers and industry associations to the Inspectors Working Group (IWG) regarding the CCS expectations:
- What is the CCS scope – viable and non-viable particulates from any origin (product residue, viruses, disinfectant residues, etc.)?”
- Should the CCS be a document, for example, a “sterility assurance quality manual” that includes an exhaustive list of expectations listed in the latest draft?
- Should manufacturers create a standalone document per site or per production buildings that describes how the strategy is applied and how the performance is measured and evaluated?
These types of questions have led the IWG to include the CCS definition in the glossary of the draft Annex 1 version 12, released in February 2020. “A planned set of controls for microorganisms, pyrogens, and particulates, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to the active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control”(1).
Despite the second revision of the draft Annex 1 v12 by IWG, it appears that the need for additional practical guidance for establishing and documenting an appropriate CCS is considered necessary by our industry. Therefore, many technical and scientific industry associations (such as PHSS/A3P, ECA, PDA, ISPE) are working actively to publish CCS practical guideline to assist the industry.
This article is part of a series of articles that will cover CCS practices, development, implementation and evaluation. This first article will discuss CCS practices and a case study of a CCS implementation. The first section presents the results of a survey conducted by STERIS to assess the scope, implementation status, and implementation needs of CCS. The second section provides a real-world example of the key elements of an existing contamination control program, and how this program was upgraded to a CCS based on a gap assessment of the existing program versus the expectations laid out in the latest EU GMP Annex 1 draft v12.
1. Contamination Control Strategy Implementation Status and Practices
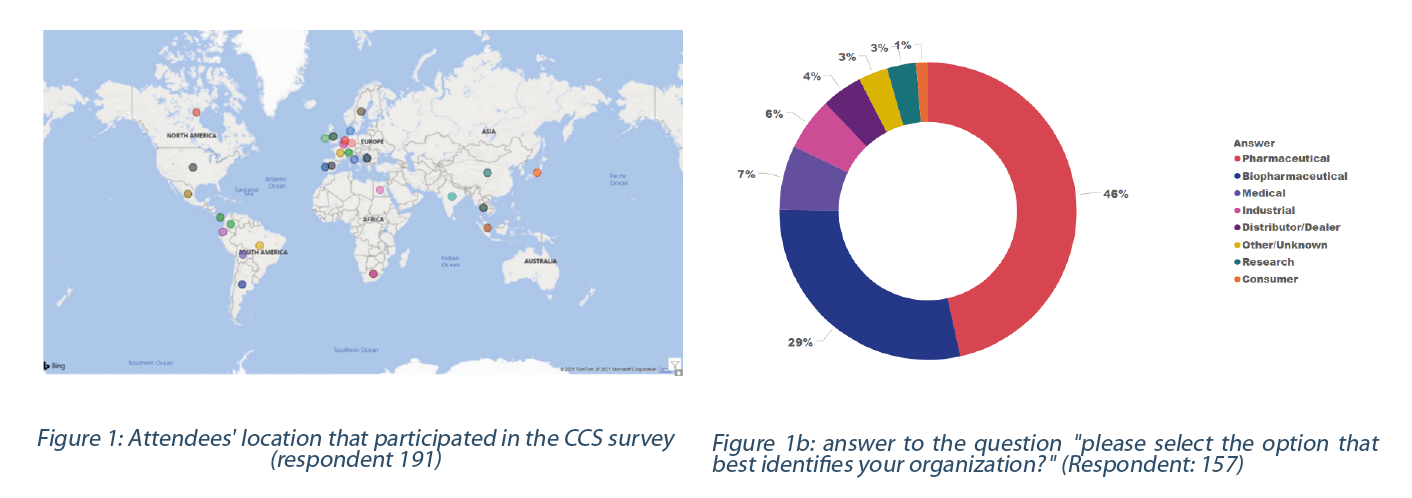
In April 2021, a survey was initiated during a STERIS hosted webinar titled “Contamination Control Strategy: Implementation Roadmap” to understand the (bio)pharmaceutical CCS implementation status, practices, and needs. One hundred ninety-one attendees located across the globe participated in the webinar (Figure 1- top and bottom). Only responses from pharmaceutical industry (i.e.: pharmaceutical, biopharmaceutical, medical, industrial, research, consumer) who responded to the survey were considered for the purpose of this analysis; the responses rate varied between 42 to 52% of the total participant.
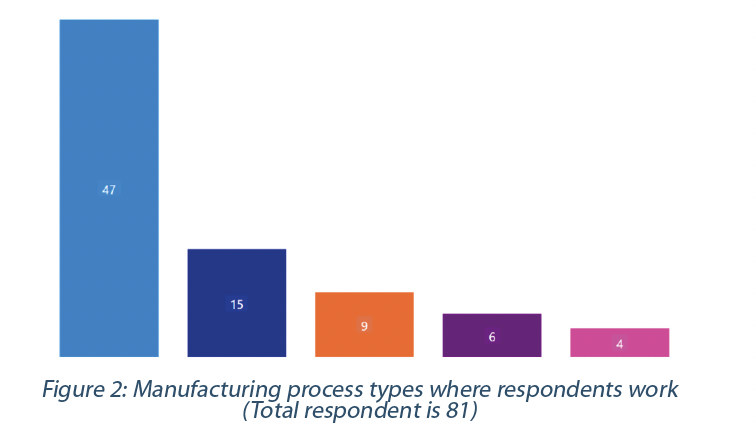
The majority of respondents work downstream (e.g., formulation, filling, and packaging) and medical devices. The remaining respondents work in upstream (e.g., bulk, drug substance manufacturing) , small molecules, and cell & gene therapy or advanced therapeutic medicinal products (Figure 2).
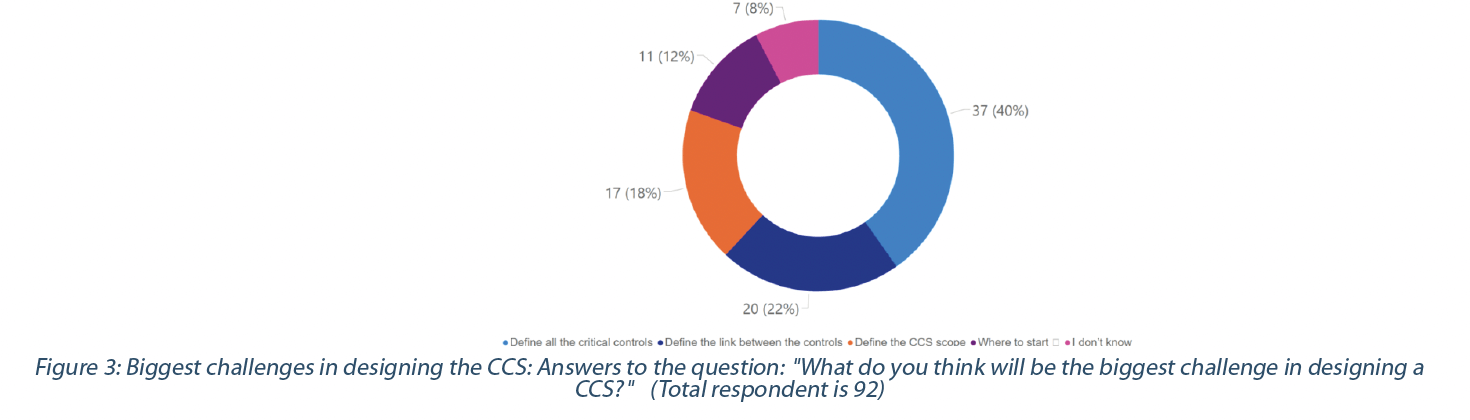
40% of the respondents see that the biggest challenge in designing a CCS is identifying all the critical controls. In comparison, 22% consider as challenging to identify the interlink between controls (Figure 3 – top). Some (18%) believe that the significant challenge is to define the CCS scope. The challenges expressed by the respondents seem to be the same across the different types of manufacturers.
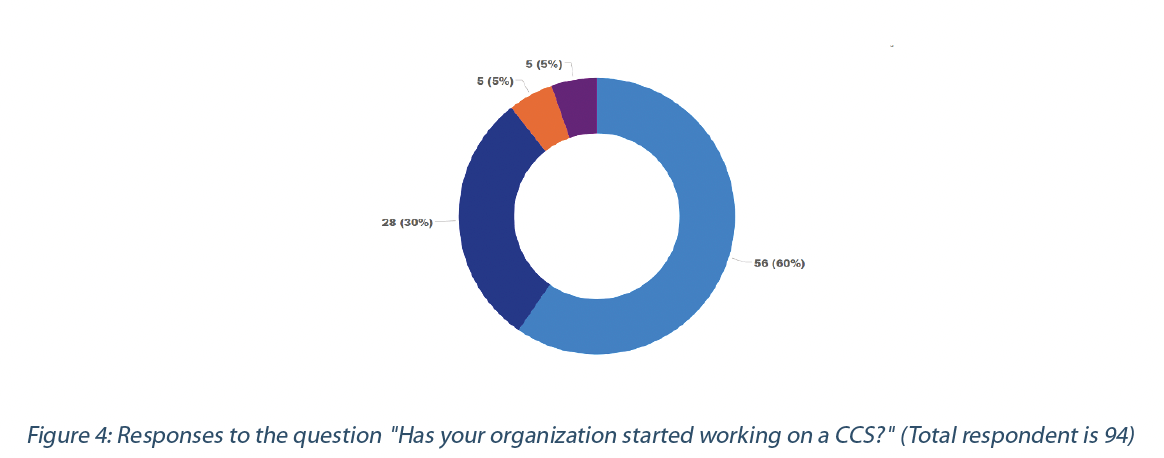
60% of the respondents are implementing a CCS, despite various challenges identified and Annex 1 being a draft. However, 29% are still in the information-gathering stage or waiting for Annex 1 to go into effect (Figure 4).
Most respondents (81% or 77) would apply the CCS to the manufacturing processes and facilities. In comparison, 7% (or 7) have also included the distribution processes where contamination may occur. The rest (12% or 11) have not defined where the CCS would be applied. It should be noted that the distribution includes transportation of intermediate or finish products between affiliates’ sites or to a contract manufacturer before or after release of the final medicinal product.
71% (or 77) have already defined the scope of their CCS, while the rest, 19% (or 18), have not yet defined it. of those who have already defined the CCS scope, 55% focus on contaminants from microorganisms, particulates, and pyrogens, while 26% focus only on microorganisms and particulates (Figure 5).
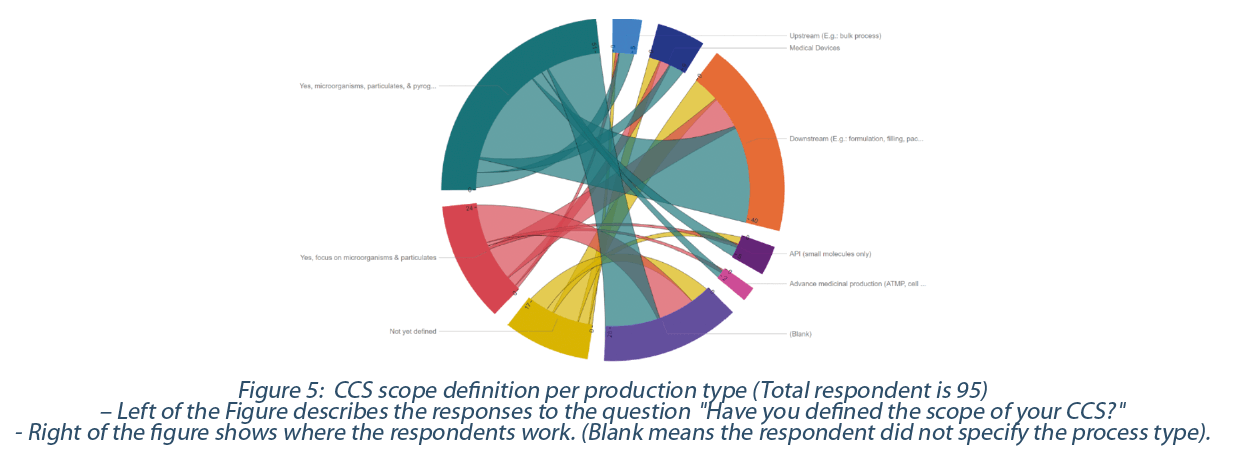
The contamination definition in the draft Annex 1 version 12 seems not to include viruses or other types of contaminations (e.g., TSE/BSE under the EMA note for guidance EMA/410/01). However, suppose viruses are identified as a source of contamination, the CCS scope should at least address this risk of contamination and how to prevent contamination.
The term particulate with respect to the CCS scope is currently subject of intense debate as some have included product residue as per EU Annex 15 in their “particulates’ ‘ definition. Therefore, they have included the concept of cross-contamination in their CCS scope. On the other hand, others argue that the definition of “contamination” in the draft Annex 1 glossary deals with foreign particulate matter (e.g visible, or sub-visible particles contamination) as expressed in the United States Pharmacopeia (USP<790>, USP<788>) or European Pharmacopeia (E.P. 2.9.20, E.P. 2.9.19). Exogeneous and environmental particulates should be included in the CCS scope. It is acceptable to have a separate document dealing with product residue or cleaning agent contamination regulated by the EU Annex 15 and Chapter 3. This last document (cross-contamination program) can be part of the genealogy of the CCS documentation. 58% (56) of respondents have included product residue in their definition of particulate, while 19% (18) have only referred to particulate as exogenous (visible and sub-visible particulates) and environmental particulates. The rest did not yet define the term particulate.
59% have or plan to have a gap assessment of their current documentation (e.g., process map, risk assessment, procedure, and organizational measures) and practices against the CCS requirements. In comparison, 10% will not perform a gap assessment (Figure 6).
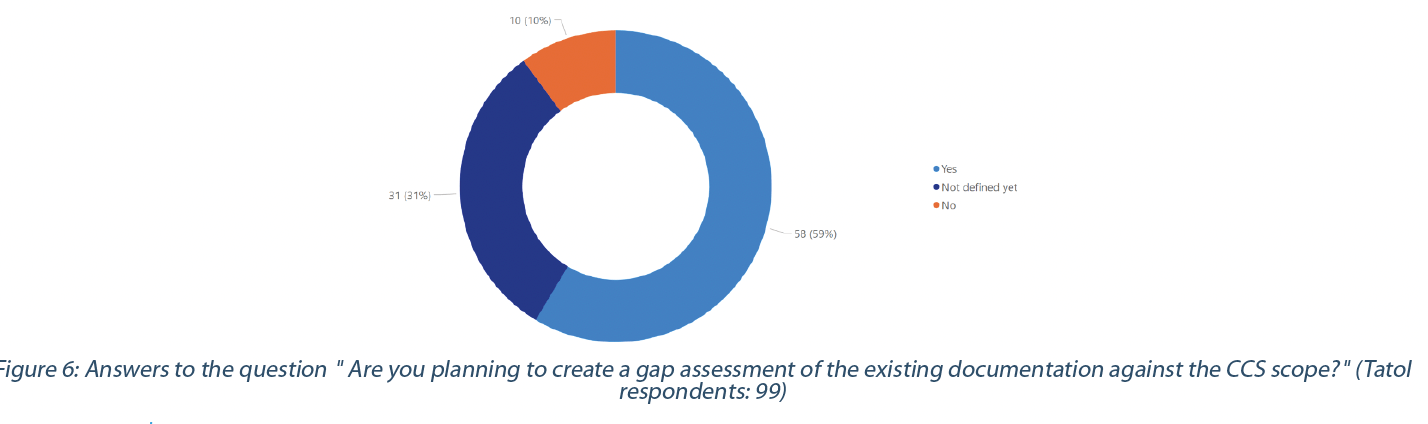
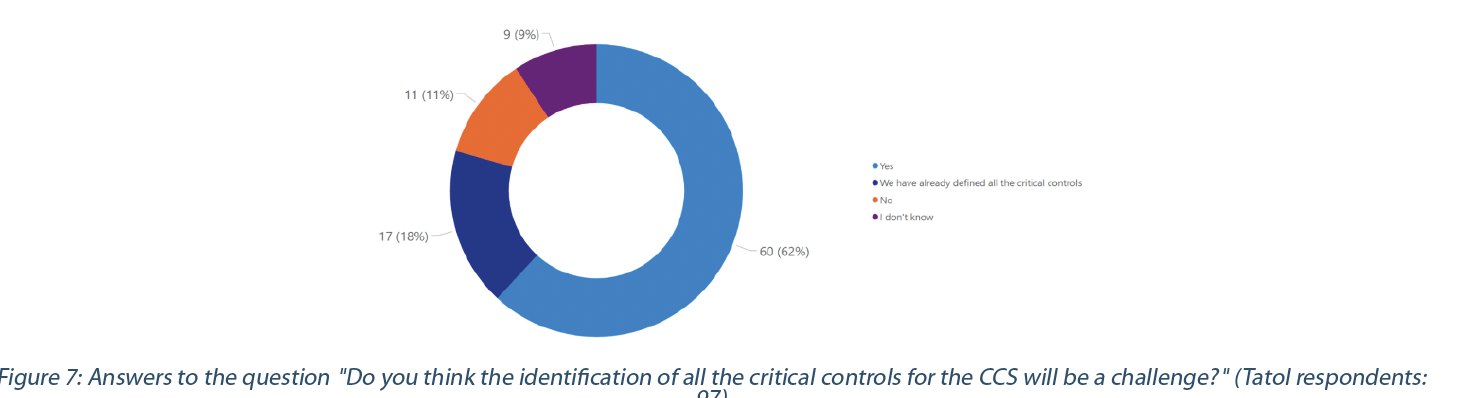
Ensuring that existing documentation and practices align with the CCS requirements and scope may help identify the critical controls. These critical controls must reflect the effectiveness of all the various controls (design, procedural, technical, and organizational) and monitoring measures employed to prevent the contamination risk. 62% of the respondents recognize the challenges in identifying all critical controls, while 11% do not think it will be a challenge. 18% of the respondents have already identified all their critical controls (Figure 7).
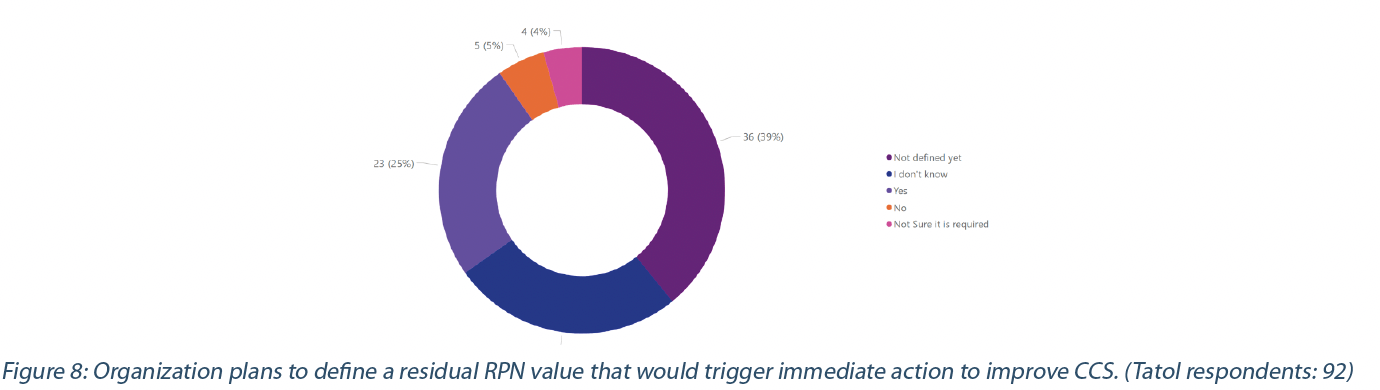
The CCS should consider all aspects of contamination control and its life cycle with ongoing and periodic review, resulting in appropriate quality system updates (1, 2). The periodic review is generally set at a defined period. However, some indicators or threshold values (e.g., Residual Risk Priority Number (RPN), quality performance indicator, Key performance indicator) suggesting an increase in the contamination risk or trend should trigger the review of the CCS to identify corrective actions(2). 39% have not defined the threshold that would trigger the CCS review, while 25% have defined it (Figure 8). Some of the respondents have not (5%) or do not think (4%) it is required to determine a threshold value; they might consider that their Pharmaceutical Quality System will trigger the corrective action to manage risks associated with contamination.
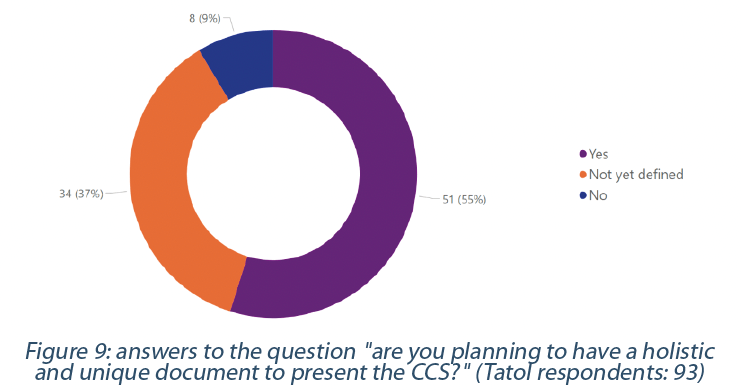
The CCS calls for a holistic view of all the critical controls involved in managing risk associated with contamination (1,2,3). This holistic view should encompass an end-to-end supply chain that includes starting material (e.g., raw material, excipient) until the final product, the facilities, and the surrounding environment (e.g., facilities, equipment). We can quickly imagine how complicated this could be with complex supply chains involving multiple entities across the globe. However, despite this complexity, 55% of respondents plan to have a holistic and unique document to present their CCS, while only 9% will not (Figure 9).
There are various ways to implement a CCS. However, a thorough understanding of the manufacturing processes and surrounding environment is required to adequately define the CCS scope and the documentation structure that fits a pharmaceutical manufacturer’s process and its supply chain complexity (2).
2. Case study of the implementation of the CCS by updating an existing Product Protection and Control Strategy (PPCS):
Disclaimer for the case study : “the gap assessment summarized in table 2 represents documentation gaps between an overarching existing PPCS approach in line with the current GMPs and applicable guidances and a newly possible CCS aligned with the requirements of the Revision 12 draft of the EU GMP Annex 1”.
Implementing a CCS requires a cross-functional team of Subject Matter Experts (SME) who have a holistic understanding of the process, facility, and product contamination risks and regulations.
The survey presented in the first section demonstrates that CCS development is a difficult exercise. It is more than just establishing a “sterility assurance quality manual.” It is about creating a strategy that includes holistic contamination risk management and ongoing evaluation of the improvement implemented to prevent contamination (1).
3. Case Study
The case study presents the update of an existing strategy called “product protection and control strategy” (PPCS) , created several years ago, that:
- Includes microbiological and particulates contamination, product residue cross-contamination, chemical containment, and product degradation. During product technology transfer to a production site, a depth risk assessment is performed to identify all critical control points at the facility and process level, followed by documentation and implementation of the PPCS.
- Is already entirely integrated into the current Product Quality Management System and part of the Site Validation Master Plan. This step provides the strategic framework that assures that the manufacturing operates in a validated state and appropriate governance to enable balanced continuous improvements and investment plans.
Step 1: verify alignment with CCS scope and documentation structure expectations versus the existing PPCS
The scope of the CCS is clarified in the definition provided in the glossary of the EU GMP Annex 1 Feb 2020 revision draft v12. In addition, in point 2.1 and point 2.5, we have the following key expectations that identify the scope further: “minimize risks of microbial, particulate, and pyrogen contamination,” and “potential sources of contamination are attributable to microbial and cellular debris (e.g., pyrogen, endotoxins) as well as particulate matter (e.g., glass and other visible and sub-visible particulates).” In terms of the CCS document structure, the latest Annex 1 revision draft does not contain any special expectation.
Therefore, based on the proposed definition and elements in the Annex 1 revision draft, the following decision has been taken as summarized in Table 1.

In conclusion of step 1: the site decided to change the name of the PPCS to CCS to align with the expected terminology while keeping the PPCS structure.
Step 2: identify the gaps between the PPCS with the new CCS requirements while maintaining the PPCS structure.
The existing PPCS considers all the aspects of the contamination control from prevention by design, control per qualification and operations, to monitoring at critical control points, sterility assurance metrics performance evaluation, and continuous improvement. This process is in good alignment with the current drafted expectations at point 2.1, 2.2, and 2.3.
The current PPCS is based on six central pillars that include several key elements that need to be described in terms of contribution to the overall contamination risk management:
A. People
- Aseptic Gown design and cleaning/sterilization qualification, related supplier qualification
- Theoretical Training and Qualification of personnel which includes in the scope: hygiene, basics elements of microbiology (contamination sources and contamination prevention measures)
- Practical qualification to gowning in aseptic areas, to sterile manufacturing operations (general cleanroom practices, aseptic techniques, product process design, disinfection)
- Participation in Aseptic Process validation
B. Facilities, Equipment, Utilities & Infrastructures Design, Qualification, Maintenance, and Control Measures
- The design sub-elements are the facility layout, cleanroom design and classification, cross-contamination management where appropriate, and people and material flow
- Airborne Contamination Control measures HEPA filtration, airflow, and pressure cascades, localized unidirectional airflow protection, airflow Pattern qualification, continuous total particulates monitoring systems where appropriate
- Biocontamination control measures any associated equipment such as cleaning, disinfection, sterilization systems/processes, and related validations
- Cleanroom’s classification, qualification, and monitoring
- HVAC, pressure cascade, utilities, and production equipment systems alarms setting and qualification – Pest control programs
- Preventive and corrective maintenance programs
- Good housekeeping programs
C. Process Design, Validation, and Control Measures
- State of the art design: using closed systems, barrier technologies, single-use systems, highly automated and integrated systems facilitating data integrity expectations, cleaning and sterilization in place, online filter integrity testing, automated decontamination systems
- Cleaning and sterilization of all equipment and primary product components
- Product sterilization by filtration validation
- Clean, dirty, aseptic, and sterile hold times qualification
- Process Control Parameters qualification and Process validation
- Manufacturing and aseptic operation activities practices qualification and monitoring
D. Product and Container Closures Design, Validation, and Control Measures
- Critical Quality Attributes determination, qualification, control, and performance monitoring
- Raw materials, excipients, container closure systems, single-use systems used in production, selection and qualification, related suppliers’ qualification, and management
- Ready to Use or Ready to Sterilize Components when more appropriate
- Container Closure Integrity Validation
- Pre-filtration Bioburden, endotoxins and other In-process Control’s validation and implementation
E. Sterility Assurance Performance Metrics and Monitoring Program
- Cleanroom Environmental monitoring data, utilities monitoring data assessment and trending
- Facility and process alarms management and trending
- Product analytical controls management, assessment, and trending (in-process, release, and stability testing data) – Raw materials, excipients, and primary products containers control and monitoring data
- Product visual inspections data (defects classification, defects levels, trending)
- Process monitoring data
- Operators’ performance monitoring and trending (personnel monitoring and qualification data)
- Aseptic Process Simulations data
- Assets Qualification Monitoring data
- Housekeeping evaluation, fit ad finish program in classified areas
- Pest control data
F. Ongoing overall Quality Oversight and Continuous Improvement
- Deviation, complaints management
- Change Management
- Site self-inspection program, quality and sterility assurance field observation, global quality audits
- Supplier management and audit program
- Management review of the quality systems and process/product performance and quality metrics
- Regulatory inspections trends and observations
- Regulatory expectations and Technological evolutions survey
The decision was made to keep the six pillars structure, as they remain aligned with the current drafted Annex 1 expectations for the CCS documentation. Potential gaps or improvements needs were identified for each key element listed in each pillar with the latest draft EU GMP Annex 1 revision text. The potential gaps identified can be improvements in procedures, processes, or points/rationale that need further clarification.
This gap assessment was performed following these steps:
- Deep line per line, word by word analysis between the newly revised draft Annex 1 v12 and the current site strategy
- A search in the regulatory framework using the following keywords: CCS, Contamination Control, Contamination.
- A benchmark of practices through external workshop, training, or seminars (e.g., A3P, ISPE, ECA, PDA workshop/training on CCS) to identify if additional elements or best practices needed to be included.
Then the new elements based on the gap assessment have been included in a template table (table 2) to facilitate the update of the existing strategy.
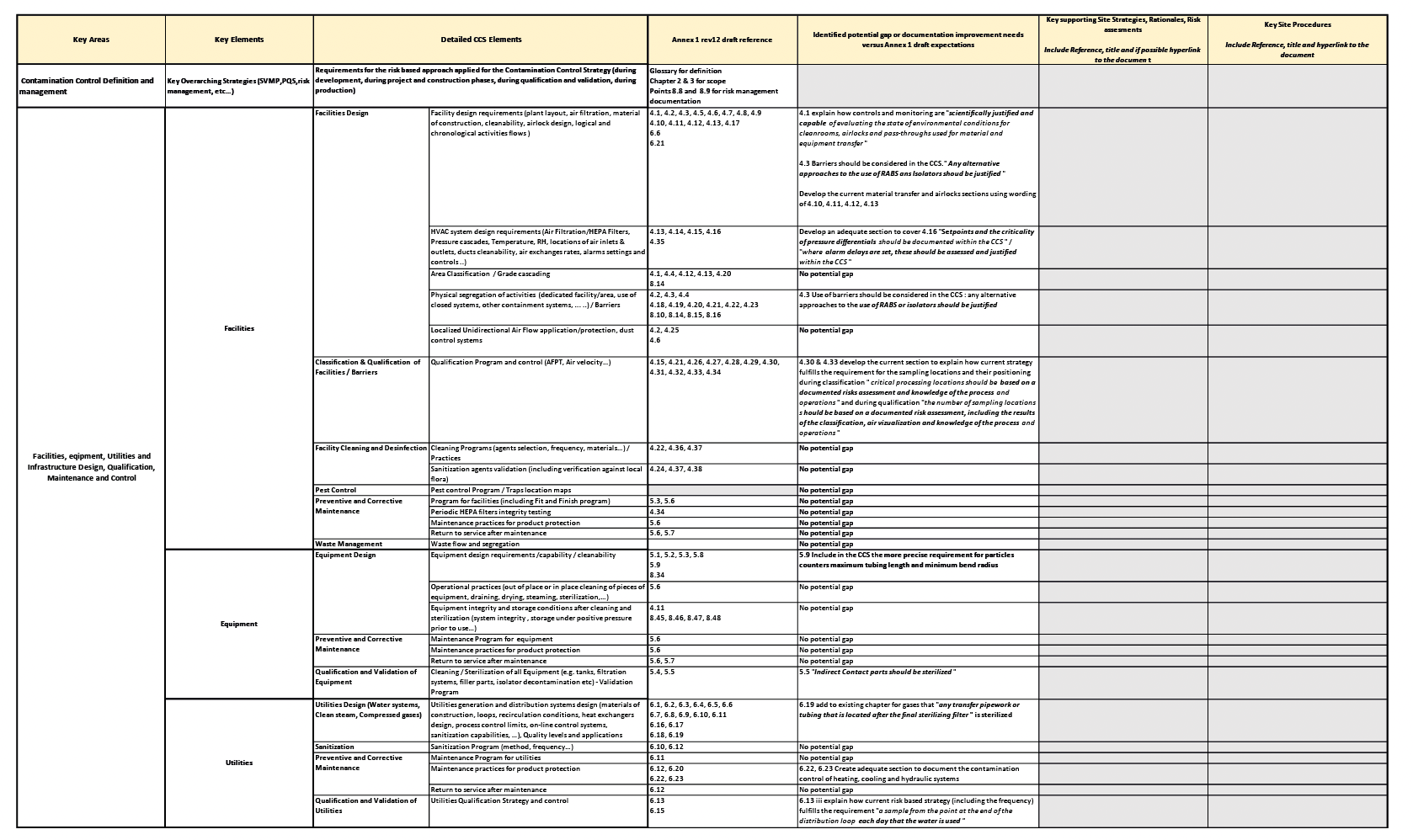
Step 3: reference all key site strategies, rationales, reports, risk assessments, procedures, plans, etc., capturing the rationale for the site’s contamination control and sterility assurance risk management program.
As described at point 3.1 of the draft Annex 1: the Pharmaceutical Quality System “PQS for sterile product manufacture should also ensure that: An effective risk management system is integrated into all areas of the product life cycle with the aim to minimize microbial contamination and to ensure the quality of the products manufactured. Risk management is applied in the development and maintenance of the CCS to identify, assess, reduce/eliminate (where applicable) and control contamination risks. Risk management should be documented and should include the rationale for decisions taken in relation to risk reduction and acceptance of residual risk. […] The risk management outcome should be reviewed regularly as part of ongoing quality management, during change control, and during the periodic product quality review.“
So far, all essential procedures were already referenced directly in the text of the documented current strategy. Still, based on the outlined new requirements, the decision was taken to collect and reference in addition the relevant key site strategies, supporting rationales, reports, risk assessments, plans, etc… to capture the justification underlining the site’s contamination control and sterility assurance risk management program. As for step 2, these new elements have to be included in the table 2 to facilitate the update section while re-using the pillars structure as presented previously.
Step 4: Update of the different sections of the CCS by the site’s Subject Matter Experts
Each site SME was involved in the gap assessment for its area of expertise and responsibilities. After the gap assessment was made, each SME had to:
- Update existing sections or create new sections to fill the gap between current procedures and regulatory/future requirements.
- Reference the adequate strategic documents, rationales, risk assessments, etc.
At the end of the four steps, the final CCS is a standalone comprehensive document with three major parts:
- The core text describes the CCS and the six pillars structure as described in step 2.
- The Table 2 records all relevant key CCS supporting strategies and assessments by the pillar as outlined in steps 3 and 4.
- A list of all Products/Production areas specific appendixes for chemical cross-contamination and product containment controls.
Step 5: Final holistic review by the CCS owner and approval by the senior management
As with any key strategic document (e.g., the site validation master plan), the CCS had to be approved by the senior management. The approvers formally commit to the described contamination risk management strategy and associated governance process to ensure product quality, patient safety, and continuous improvements by approving this CCS.
It is clear that this CCS document will become a major expected deliverable of a sterile products manufacturing site during future regulatory inspections. This document includes a complete overview of contamination control management. It can directly be used as a support for the inspectors to verify the effective application of the described process and controls and to assess the robustness of the referenced supporting rationales and risks assessments.
It must be acknowledged that the creation (and even the update) of such a comprehensive document represents a challenge and necessitates an excellent transverse knowledge of the process and facility control elements. On the other hand, once finalized and approved, this CCS is also an excellent training tool for all employees who must be aware of their role in the holistic contamination control picture.
One of the biggest challenges foreseen is defining an effective transversal performance dashboard and governance decision-making as suggested by the draft Annex 1 at point 2.4.
4. Conclusion
The CCS is a key strategic document/plan that describes the contamination risk management strategy and associated governance to decide the continuous improvements and investment plans to prevent contamination. Therefore, developing such a document requires a cross-functional team of experts with good production, QRM, and regulatory knowledge. This work will undoubtedly require extensive hours of meetings and teamwork.
Despite the second revision of the draft Annex 1 by Inspector Working Group (IWG) and several industry conferences and workshops around the CCS topic, need for additional practical guidance for establishing and documentation of an appropriate CCS is considered necessary by our industry.
It is logical to encounter different CCS understanding and implementation practices as suggested by the survey presented. This phenomenon may explain why the CCS scope may be different between manufacturers based on:
- the type of processes (e.g., downstream, upstream, medical device),
- type of product manufactured (sterile, non-sterile),
- manufacturer process knowledge and expertise,
- understanding of the draft Annex 1 version 12,
- the existing contamination control program in place.
Consequently, the implementation and the evaluation of the CCS will also differ between manufacturers. The practice difference is acceptable when the manufacturer can justify that the CCS will comply with regulatory requirements.
This article shared one example of defining the CCS scope and implementing a CCS by updating an existing contamination control program.
However, it is up to the manufacturer to decide the scope and the elements (also called “points to consider” in draft Annex 1) being part of the CCS to create a strategy that includes a holistic contamination risk management and ongoing evaluation of the improvement implemented to prevent contamination.
The survey and the case study shed light on the industry challenges in:
- Agreeing on the CCS scope,
- Identifying all the critical controls,
- Organizing people roles and the PQS around the CCS, e.g., creating a new role, creating new governance bodies, or integrate it within the existing organization,
- Defining the threshold that would require review/evaluation of the CCS,
- Developing an effective transversal quality performance CCS dashboard.
This article is part of a series of articles. The second article will focus on the challenges of developing and implementing a CCS during the early design phase of a new project. The third article will present an example of an evaluation program to assess the performance of the CCS and how an efficient continuous improvement plan is identified.
Share


Isabelle Hoenen
hoenen_isabelle@lilly.com

Walid El Azab
Walid_ElAzab@steris.com
References
[1] : Second targeted stakeholders’ consultation on the draft revision 12 of Annex 1, on manufacturing of sterile medicinal products, of Eudralex volume 4 – Public Health – European Commission. https://ec.europa.eu/health/medicinal_products/consultations/2020_sterile_medicinal_products_en (accessed Jun 10, 2020)
[2] : Walid El Azab, Contamination Control Strategy: Implementation Roadmap, PDA Journal of Pharmaceutical Science and Technology, Volume 75, Number 5, September/October, 2021.
[3] : Johnson, L.; Hansy, C. Establishing a Contamination Control Strategy/Program: From Global Development to Site Implementation. https://www.americanpharmaceuticalreview.com/Featured-Articles/564173- Establishing-a-Contamination-Control-Strategy-Program-From-Global-Development-to-Site-Implementation/?catid=6262 (accessed Jun 10, 2020).
