
Summary
- Viewpoint of the ANSM’s Inspection Division (DI) concerning the ICH Q12 guideline
- ICH Q12: the basics. Review of the work of the A3P ICH Q12 GIC
- First steps towards ICH Q12: Leveraging process understanding & development data to define process Established Conditions
- ICHQ12 Implementation from an Industry Perspective with a Focus on Established Conditions
- ICH Q12 compliance and Unified Quality and Regulatory Information Management
- Burkholderia cepacia strikes again
- New EMA Sterilization guideline Guideline on the sterilisation of the medical product, active substance, excipient and primary container (EMA/CHMP/CVMP/QWP/850374/2015)
- How to store highly sensitive drugs? The benefit of functional coatings
ICH Q12: the basics. Review of the work of the A3P ICH Q12 GIC
The ICH Q12 guideline, “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management”[1]published in draft version in November 2017, led to the creation of an A3P common interest group (GIC) in June 2018. Bringing together around twenty health industry experts (Regulatory, CMC, Quality Assurance, Validation experts, etc.), the objective of the GIC was to comment on the draft guideline submitted for public consultation for a period of 1 year, until the end of 2018.

In 2019, the ICH set up an Expert Working Group (“Expert Working Group”), dedicated to discussions about the comments arising from the consultation phase and the revision of certain parts of the text. During the Singapore assembly at the end of November 2019, the final guideline was adopted (“Step 4”). The final version was eagerly awaited by industry in view of the considerable challenges involved in harmonization and the facilitation of post-approval change management.
The adjustments made to the text between the draft version and the final version are in the process of being analyzed by the stakeholders. In the next milestones in the implementation phase (“Step 5”), the ICH announced the launch of training activities in spring 2020.
The ICH harmonization process is recalled in Figure 2, with the main milestones given by the ICH for Q12.

1. The objectives of Q12
The main objective of the ICH Q12 guideline is to provide a framework for the management of post-approval changes with an impact on the Quality part (Module 3/CMC) of the CTD. Its purpose is to facilitate the management of post-approval changes and to make the process for handling these more predictable and efficient, enabling industry to manage a large proportion of their changes via a pharmaceutical quality system in order to minimize regulatory variations. The opportunities envisaged due to the implementation of Q12 depend on the application of science and risk-based approaches (QbD ou Science&Risk based approach / cf. ICH Q8 et Q9) and a robust Pharmaceutical Quality System (cf. ICH Q10). Hence, this first ICH guideline specifically dedicated to the post-approval phase of the product lifecycle (commercial phase) is in line with the existing Q8, Q9, Q10 and Q11 guidelines, the links with these being shown in Figure 1.
In a context of transparency between industry and regulatory authorities, the stated objective of the ICH Q12 guideline is to support innovation and continual improvement.
2. 2. Current situation for “Product Lifecycle Management” and challenges
A number of regulatory texts currently govern post-approval change management, with the rules being set by the regulatory authorities of the regions concerned. The regulations currently in force in Europe[3], the USA[5,6], or the WHO[4] (for biological products) already enable approaches for the categorization of changes according to risk. Depending on their impacts on Product requirements (Quality, Safety and Efficacy), changes are categorized and submission procedures are adapted (examples in Figure 3).

At present, post-approval change management rules are not harmonized around the world. A product registered in the USA, Europe, Japan, Brazil or China, etc. will, for the same change, follow different and specific submission procedures. Depending on each authority, specific data must be provided to support the change in order to address certain specific requirements of the authorities. The implementation times can be very variable for the same change (example of approval times for the same change in Figure 4), ), making it necessary to manage transitional periods of several years before full industrial implementation.
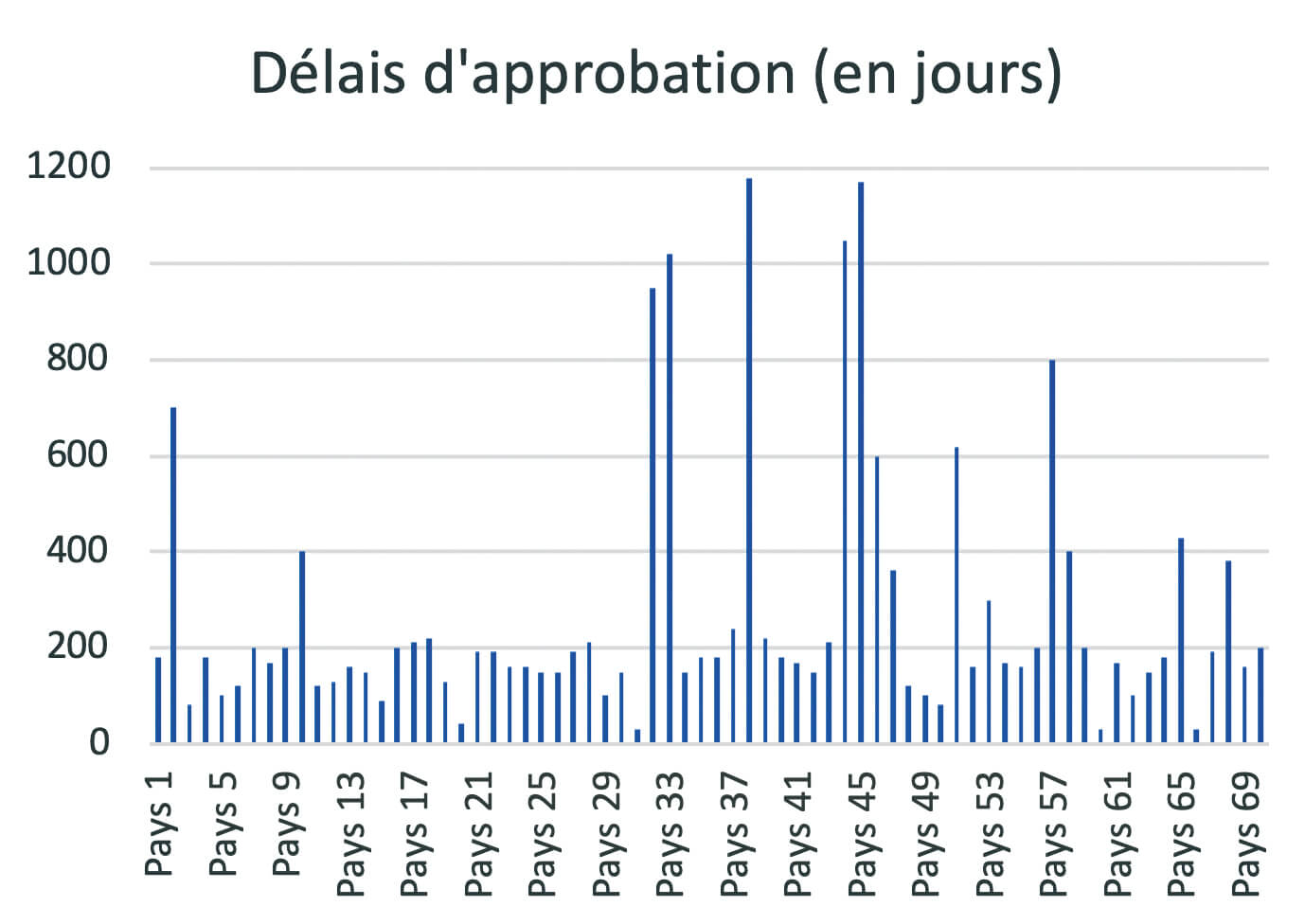
The consequences of these regional specificities can be an obstacle to process improvement and product quality, result in risks of error or stock-out and lead to substantial additional costs.
There is a strong demand on the part of industry for the harmonization of these post-approval change management procedures. The objective is to achieve alignment in terms of the data that needs to be provided and more predictable implementation times and to be able to more systematically apply a common approach concerning risk-based change categorization to focus efforts on the changes with the greatest impact.
The challenges of reducing the number of regulatory submissions, optimizing implementation times, reducing workload and the associated costs are such that numerous industry players are already preparing to implement the concepts and tools described in ICH Q12.
3. Content of ICH Q12
The text introduces or reinforces tools and enablers intended to link different phases of the product lifecycle.
1. 1. Categorization of changes : still risk-based, to guide, depending on the changes, the type of communication between the MA holder (Marketing Authorization Holder or MAH) and the regulatory authorities.
2. Established Conditions (EC) : are described as the elements to assure Product Quality, characterize the commitments defined in the CTD. They require a regulatory variation if changed.
3. Post-Approval Change Management Protocol (PACMP) : also called “Comparability Protocol” in some regions and already used. This defines the requirements and studies that need to be implemented in the event of post-approval changes. Once this protocol has been approved by the regulatory authorities, the change may be envisaged with greater predictability.
4. Product Lifecycle Management (PLCM) document : serves as a central framework for product lifecycle management, to capture ECs and the essential elements of the Control Strategy. It is described as a new tool to communicate to the regulatory authorities the management envisaged for Post-approval changes, and should be seen as a living document that may evolve on the basis of evolutions in Control Strategy and Post-approval changes.
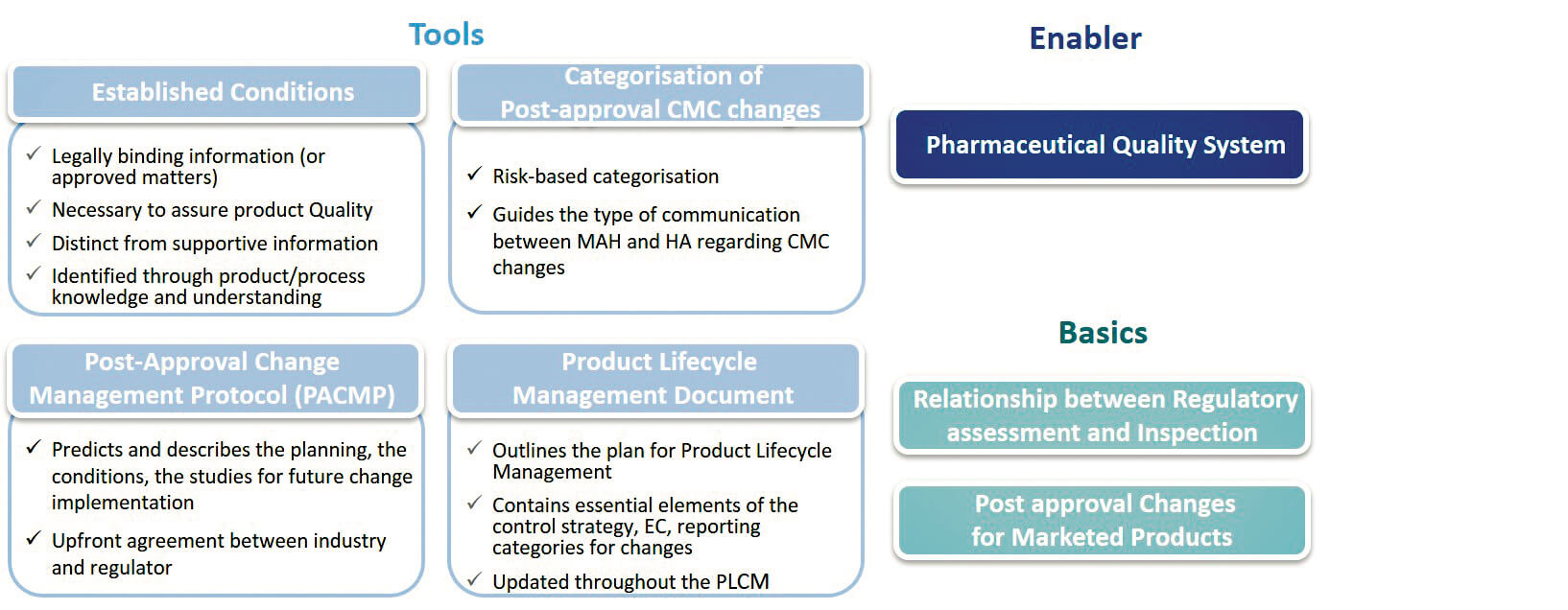
The guideline highlights essential requirements, such as the PQS and Change Management according to the ICH Q10 vision, and recalls the basics, such as the relationship between the two activities of the regulatory authorities – assessment and inspection -, for which the close relationship and complementary roles are stressed. The guideline also paves the way for allowing marketed products (Legacy Products) to also benefit from the application of ECs, PACMPs.
4. A3P GIC comments
The draft version of the guideline published in 2017 was therefore submitted for a consultation process over the course of 2018. A3P’s comments, sent to the EMA in December 2018, were compiled by the EMA and published in January 2019, along with those of 11 other stakeholders, in a document entitled “Overview of comments received on ICH guideline Q12 on technical and regulatory considerations for pharmaceutical product lifecycle management (EMA/CHMP/ ICH/804273/2017)”[2].
A total of 59 comments and changes were proposed by the A3P Group, including 4 general and 55 specific comments. The specific comments can be broken down as follows (see Figure 6), depending on the chapters, appendices and annexes to the text.

All the GIC A3P comments are available in the document published by the EMA [2]. Overall, the A3P Group fully supports the concepts in the ICH Q12 guideline.
The Group expressed one major concern concerning international harmonization. Since the regulatory provisions of the different ICH regions are not fully compatible with the requirements introduced in the ICH Q12 guideline, industry players issue some warnings concerning these points: if the regional specificities and disparities were to continue, the benefits of ICH Q12 would be very limited and its objectives would not be met.
One chapter of the guideline prompted numerous comments: this relates to the “Established Conditions”, which need to be better defined in the Group’s view. Concepts such as “implicit” and “explicit” were introduced and called for clarifications. The concepts of “implicit” and “explicit” have been removed from the final guideline. A decision tree for identification of ECs was provided in the draft guideline and required a few adaptations for greater clarity. The decision tree has been reworked and a new version is available in the final version. The concept of KPP (Key Process Parameters) was introduced in the draft version of this ICH guideline. It proposed a definition that raised numerous questions. The term has been removed from the final guideline, suggesting that no consensus was obtained with respect to its definition.
Finally, the A3P Group proposes reinforcing the notion of Control Strategy and specifying its content: the Control strategy includes Established Conditions and non Established Conditions.
The chapter on the Post Approval Change Management Protocol (PACMP) mainly gave rise to requests for clarification or suggestions on the part of the Group to include it in certain parts of the regulatory application. The Group would like to specify that it may be included in an initial application or may be submitted subsequently as a standalone document.
The Product Lifecycle Management Document, a new tool introduced in Q12 gave rise to numerous debates and opinions. An ICH recommendation with respect to the location in the CTD was requested by the Group, which suggested Module 1 (global overview in Module 1) or Module 3 (scientific analysis, technical justifications). The final version of the guideline suggests module 3.2.R or Module 1.
Numerous questions were raised concerning the coexistence of the PLCM/Q12 approach and the traditional Change management and Variations approach. How will the PLCM document be managed? How to monitor the changes implemented via PQS? All these concrete and operational questions will be raised in the coming years during the Q12 implementation phase. Industry is well aware of the challenges that will need to be faced, such as the implementation of this approach for products that are already marketed, developed using traditional approaches, for which substantial efforts will be necessary to identify the Established Conditions, prepare and submit the PLCM, etc. Pragmatic priority-setting will be necessary, informed by the long-term challenges and contributions, on the basis of the lifecycle of each product.
The inclusion of examples in the annexes to the ICHQ12 guideline to explain the text’s concepts was much appreciated by the Group. Since the guideline has a broad scope in terms of product types, examples such as sterile products and aseptic filling, would be useful to supplement illustration of concepts. In the final version, additional examples have been provided, in particular via a new annex concerning the identification of Established Conditions for analytical methods.
Finally, specifying the CTD sections in which the different tools and documents are expected (e.g. PLCM document) will enable more effective harmonization in ICH regions.
As concerns the comments of industry and other associations compiled by the EMA, they are globally consistent for the common parts of the text. The common general comment concerns harmonization with the “regional” regulatory framework. The importance of reworking the robustness of the Quality Management System is highlighted by all.
5. Conclusions and prospects
2019 was a year dedicated to review of the comments submitted by industry and associations. ICH Expert Working Group (EWG) sessions led to adoption of the final text on November 20, 2019. Industry is currently discovering the final version of the guideline and the choices that have been made by the ICH in the implementation of the comments. There is no doubt that significant harmonization work is needed before the guideline can be effectively and efficiently implemented. This will require industry and the regulatory authorities to work together in order to make the concepts operational and agree on their interpretation. The FDA has already initiated this exchange process, launching a pilot program in 2019, in which pharmaceutical companies submitting an application for assessment were able to propose Established Conditions and benefit from a review with the FDA.
The exchanges over the few months of the A3P GIP’s work resulted in consideration that the scientific knowledge and risk-based QbD approaches will enable increases in expected financial savings, authorizing Changes historically subject to approval to be implemented under the oversight of the QMS. This vision, which encourages innovation and continual improvement, therefore requires an extremely robust QMS across the entire medicinal product manufacturing chain and operational Knowledge Management in order to value all the Product/Process understanding acquired during development and supplemented throughout the Product lifecycle.
Share article


Johanne Piriou – AKTEHOM
Johanne Piriou, Biochemistry / Biotechnology engineer (INSA Lyon), has been a consultant in Product / Pharmaceutical Process control at AKTEHOM.
JJohanne has carried out commissioning, qualification, validation and start-up projects for production units (Sterile finished products or Drug Substance intended for sterile finished products).
Through these installation start-up projects and by supporting continuous improvement and remediation plans, Johanne specializes in controlling microbiological contamination and ensuring sterility. Johanne’s areas of expertise complement each other with subjects such as the operational implementation of the QbD approach, the definition of Control Strategy and their implementation, as well as Process Validation.
Today, Johanne oversees Control Strategy, QbD, and Product Lifecycle Management expertise within the consultancy firm AKTEHOM.
Bibliography
(1) ICH Q12 – Technical and regulatory considerations for pharmaceutical product lifecycle management – Step 2 – Novembre 2017.
(2) Overview of comments received on ICH guideline Q12 on technical and regulatory considerations for pharmaceutical product lifecycle management (EMA/CHMP/ICH/804273/2017)
(3) Commission Regulation 1234/2008
(4) WHO/OMS – Guidelines on procedures and data requirements for changes to approved biotherapeutic products – WHO/PAC for BTPs_DRAFT/2016.
(5) Guidance for Industry – Changes to an Approved NDA or ANDA (2004)
(6) Guidance for Industry – CMC Postapproval Manufacturing Changes To Be Documented in Annual Reports (2014)
(7) FDA – GFI – Established Conditions: Reportable CMC Changes for Approved Drug and Biologic Products – Draft 2015.
Glossary
AMM : Autorisation de Mise sur le Marché
CMC : Chemistry, Manufacturing, Control
CTD : Common Technical Document
EC : Established Condition
EMA : European Medicines Agency
EWG : Expert Working Group
GIC : Groupement d’Intérêt Commun
ICH : International Conference of Harmonization
KPP : Key Process Parameter
PACMP : Post Approval Change Management Protocol
PLCM : Product Life Cycle Management
PQS : Pharmaceutical Quality System
QbD : Quality by Design
QMS : Quality Management System
