


Sommaire
- Point de vue de la direction de l’inspection (DI) de l’ANSM sur le document ICH Q12
- ICH Q12 : les fondamentaux. Retours des travaux du GIC A3P ICH Q12
- First steps towards ICH Q12: Leveraging process understanding & development data to define process Established Conditions
- ICHQ12 Implementation from an Industry Perspective with a Focus on Established Conditions
- ICH Q12 compliance and Unified Quality and Regulatory Information Management
- Burkholderia cepacia a encore frappé
- Nouveau guideline Stérilisation de l’EMA
- How to store highly sensitive drugs? The benefit of functional coatings
ICH Q12 : les fondamentaux. Retours des travaux du GIC A3P ICH Q12
Le guide ICH Q12, “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management”[1], publié en version draft en Novembre 2017, a déclenché la constitution d’un GIC A3P en Juin 2018. Constitué d’une vingtaine d’experts de l’industrie de la santé (experts Réglementaires, CMC, Assurance Qualité, Validation…), l’objectif du GIC était de commenter le texte, soumis à consultation publique durant 1 an, jusque fin 2018.

En 2019, l’ICH a constitué un Groupe d’experts (“Expert Working Group”), dédié aux discussions sur les commentaires issus de la phase de consultation et à la reprise de certains éléments du texte. Lors de la session de Singapour fin Novembre 2019, le guide final a été adopté (“Step 4”). La version définitive était très attendue par les industriels au vu des enjeux considérables que représente une harmonisation et une facilitation de la gestion des changements post approbation.
Les ajustements du texte entre la version draft et la version finale sont en cours d’analyse par les parties prenantes. Dans les prochains jalons de la phase d’implémentation (“Step 5”), l’ICH a annoncé le lancement des activités de formation au printemps 2020.
Le processus d’harmonisation ICH est rappelé en Figure 2 avec les principaux jalons donnés par l’ICH pour le Q12.

1. Les objectifs du Q12
Le guide ICH Q12 a pour objectif principal d’établir un cadre pour la gestion des changements post-approbation qui impactent la partie Qualité (Module 3/CMC) du dossier CTD. Sa finalité est de faciliter la gestion des changements post-approbation, de rendre leur processus de traitement plus prévisible et efficace, en permettant aux industriels de gérer une grande partie de leurs changements au travers du système qualité pharmaceutique afin de minimiser les variations réglementaires. Les opportunités envisagées grâce à l’implémentation du Q12 sont conditionnées par la mise en place des démarches basées sur la connaissance scientifique et le risque (QbD ou Science&Risk based approach / cf. ICH Q8 et Q9) et la mise en place d’un Système de Management de la Qualité robuste (cf. ICH Q10). Ainsi, ce premier guide ICH spécifiquement dédié à la phase post-approbation du cycle de vie du produit (phase d’exploitation commerciale) s’inscrit en continuité des textes existants Q8, Q9, Q10 et Q11, dont l’articulation est représentée en Figure 1.
Dans un contexte de transparence entre l’industrie et les autorités de réglementation, le but affiché du guide ICH Q12 est de faciliter l’innovation et à la mise en œuvre de l’amélioration continue.
2. Situation actuelle du “Product Lifecycle Management” (Gestion du cycle de vie du produit) et enjeux
Un certain nombre de textes réglementaires régissent actuellement la gestion des changements post-approbation, les règles étant fixées par les autorités réglementaires des régions concernées. Les textes en vigueur actuellement en Europe[3], aux USA[5,6], ou encore l’OMS[4] (pour les produits biologiques) permettent déjà les approches de catégorisation des changements par le risque. En fonction de leurs impacts sur les requis du Produit (Qualité, Sécurité et Efficacité), les changements sont catégorisés et les procédures de soumission sont adaptées (exemples en Figure 3).

Aujourd’hui, les règles de gestion des changements post approbation ne sont pas harmonisées à travers le monde. Un produit enregistré aux USA, en Europe, au Japon, au Brésil, en Chine… suivra, pour le même changement, des procédures de soumission différentes et spécifiques. Selon chacune des autorités, des données spécifiques devront être fournies pour supporter le changement afin de répondre à certains requis spécifiques des autorités. Les délais d’implémentation peuvent être très variables pour un même changement (exemple de délais d’approbation pour un même changement en Figure 4), ce qui amène à devoir gérer des périodes de transition de plusieurs années avant une mise en œuvre complète au niveau industriel.
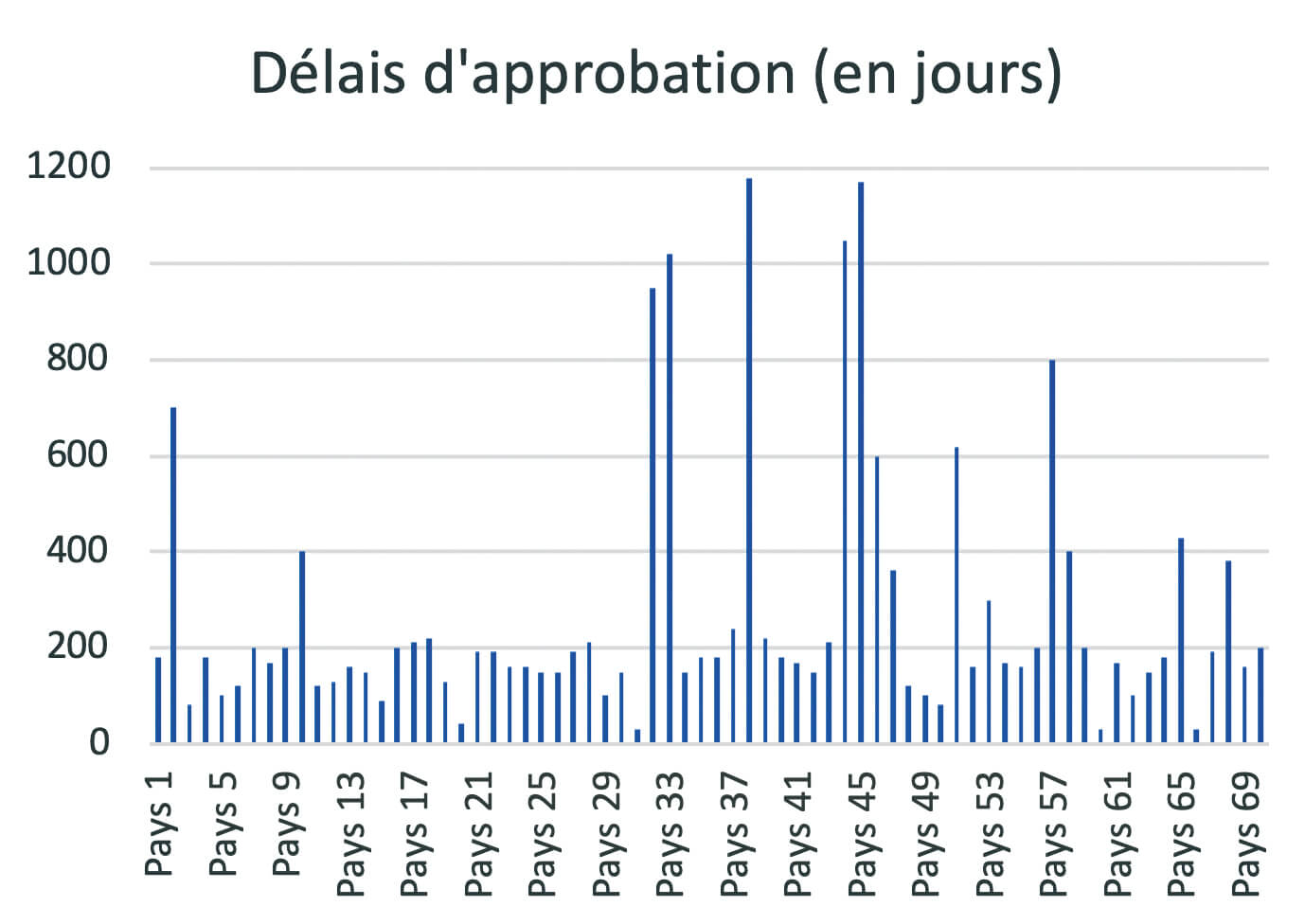
Les conséquences de ces spécificités régionales peuvent représenter un frein à l’amélioration des procédés et la qualité des produits, des risques d’erreur et de rupture, et des coûts additionnels conséquents.
L’industrie est en forte attente d’une harmonisation de ces procédures de gestion des changements post approbation. L’objectif est d’obtenir l’alignement sur les exigences de données à fournir, des durées d’implémentation plus prévisibles, et de pouvoir plus systématiquement appliquer une approche commune concernant la catégorisation des changements basée sur le risque pour focaliser les efforts sur les changements les plus impactant.
Les enjeux de réduire le nombre des soumissions réglementaires, optimiser les délais d’implémentation, réduire la charge de travail et les coûts associés sont tels que de nombreux industriels se préparent déjà à la mise en œuvre des concepts et outils décrits dans l’ICH Q12.
3. Contenu de l’ICH Q12
Le texte introduit ou renforce des outils et facilitateurs qui doivent permettre de lier les différentes phases du cycle de vie.
1. Catégorisation des changements : toujours basée sur le risque, pour orienter, en fonction des changements, le type de communication entre le détenteur de l’AMM (Titulaire de l’Autorisation de Mise sur le Marché) et les autorités réglementaires
2. Established Conditions (EC) : sont décrites comme les requis garantissant la Qualité du produit, caractérisent les engagements définis dans le CTD. Elles exigent une variation réglementaire si modifiées
3. Post-Approval Change Management Protocol (PACMP) : aussi appelé “Comparability Protocol” dans certaines régions et déjà utilisé. Il définit les exigences et les études nécessaires qui devront être mises en œuvre lors d’un changement post-autorisation. Une fois l’accord obtenu des autorités réglementaires sur ce protocole, le changement pourra être envisagé avec davantage de prévisibilité.
4. Product Lifecycle Management (PLCM) document : représente un référentiel central pour la Gestion du cycle de vie du produit, pour capturer les ECs et les éléments essentiels de la Control Strategy. Il est décrit comme un nouvel outil pour communiquer aux autorités réglementaires la gestion envisagée pour les Changements post-approbation, et devrait être un document vivant qui pourra évoluer en fonction de l’évolution de la Control Strategy, des Changements post-approbation.
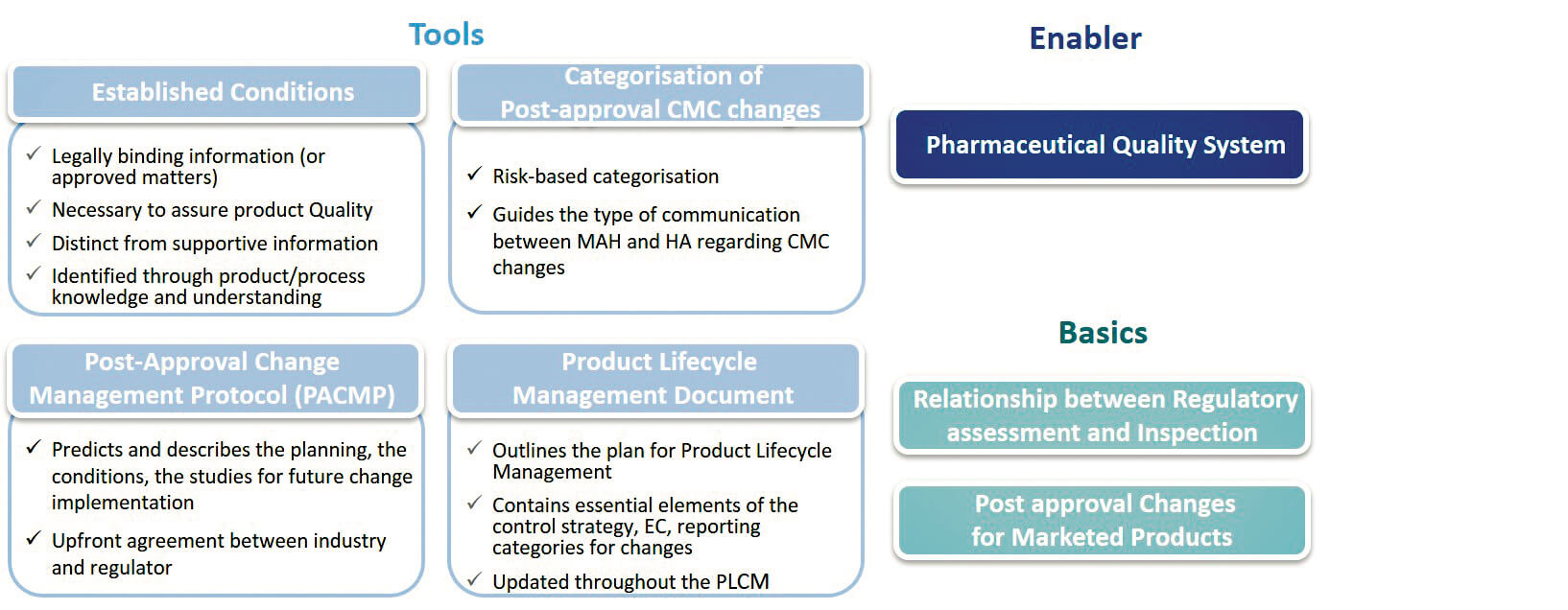
Le guide met en avant des requis essentiels tels que le PQS et le Change management selon la vision ICH Q10, et rappelle des fondamentaux tels que la relation entre les deux volets des autorités réglementaires, l’évaluation et l’inspection, dont la relation étroite et le rôle complémentaire sont soulignés. Le guide ouvre la possibilité de faire bénéficier également aux produits commercialisés (Legacy Products) l’application des EC, PACMP.
4. Commentaires du GIC A3P
La version draft du guide publiée en novembre 2017 a donc été soumise à consultation durant l’année 2018. Les commentaires de l’A3P, envoyés à l’EMA en Décembre 2018, ont été compilés par l’EMA et publiés en Janvier 2019, avec ceux de 11 autres parties prenantes dans un document intitulé “Overview of comments received on ICH guideline Q12 on technical and regulatory considerations for pharmaceutical product lifecycle management (EMA/CHMP/ICH/804273/2017)”[2].
Au total, 59 commentaires et changements ont été proposés par le Groupe A3P, dont 4 généraux et 55 commentaires spécifiques. Les commentaires spécifiques se répartissent de la façon suivante (voir Figure 6), selon les chapitres, appendices et annexes du texte.

L’ensemble des commentaires du GIC A3P est disponible dans le document publié par l’EMA [2]. Globalement, le Groupe A3P supporte pleinement les concepts du guide ICH Q12.
Le Groupe a émis une préoccupation majeure au niveau de l’harmonisation internationale. Le corpus réglementaire des différentes régions ICH n’étant pas parfaitement compatible avec les requis introduits par le guide ICH Q12, les industriels alertent sur ces points : si les spécificités et considérations régionales perduraient, les bénéfices de l’ICH Q12 seraient très limités et son objectif non atteint.
Un chapitre du guide a été très commenté : il concerne les “Established Conditions”, qui nécessitent d’être mieux définies selon le Groupe. Des notions telles que “implicite” et “explicite” étaient introduites et appelaient des précisions. Ces notions “implicite” et “explicite” ont été retirées du guide définitif. Un arbre décisionnel permettant d’identifier les ECs était fourni dans le guide draft et demandait quelques adaptations pour plus de clarté. Le logigramme a été retravaillé et une nouvelle version est disponible dans la version finale. Le concept des KPP (Key Process Parameters) était introduit dans cet ICH en version draft. Il proposait une définition posant de nombreuses questions. Le terme a été retiré du guide définitif, laissant présumer que le consensus n’a pas été obtenu sur sa définition.
Enfin le Groupe A3P propose de renforcer la notion de Control Strategy et de préciser son contenu : la Control strategy contient des Established Conditions et des non Established Conditions.
Le chapitre sur le Post Approval Change Management Protocol (PACMP) a essentiellement appelé des demandes de précisions ou des suggestions de la part du Groupe pour l’inclure dans certaines parties du dossier réglementaire. Le Groupe souhaiterait faire préciser qu’il peut être soumis lors du dépôt initial ou plus tard, comme un document autonome pouvant être soumis indépendamment.
Le Product Lifecycle Management Document, nouvel outil introduit dans le Q12 a suscité de nombreux débats et prises de position. Une recommandation de l’ICH sur la localisation dans le CTD était souhaitée par le Groupe, qui suggérait le Module 1 (global overview dans le Module 1) ou le Module 3 (données scientifiques, justifications techniques). La version finale du guide propose le module 3.2.R ou le Module 1.
De nombreux questionnements sont apparus à propos de la coexistence de l’approche PLCM/Q12 et de l’approche historique de la gestion des Changements et Variations. Comment le PLCM document va-t-il être géré ? Comment suivre les changements implémentés via le PQS ? Autant de questions concrètes et opérationnelles qui se poseront dans les prochaines années lors de la phase d’implémentation du Q12. Les industriels sont conscients des challenges qui se présentent tels que la mise en œuvre de cette démarche sur les produits déjà commercialisés, développés selon des approches traditionnelles, pour lesquels des efforts conséquents seront nécessaires pour identifier les Established Conditions, préparer et soumettre le PLCM… Des priorisations devront être réalisées avec pragmatisme, éclairées par les enjeux et apports sur le long terme, en fonction du cycle de vie de chaque produit.
L’intégration d’exemples en annexe du guide ICHQ12 a été appréciée par le Groupe pour expliciter les notions du texte. Le guide ayant un périmètre large en termes de typologies de produits, des exemples tels que des produits stériles et procédés aseptiques seraient utiles pour compléter l’illustration des concepts. Dans la version finale, les exemples ont été complétés, notamment par une nouvelle annexe concernant l’identification des Established Conditions pour les méthodes analytiques.
Enfin, préciser les sections du CTD dans lesquelles les différents outils et documents sont attendus (e.g. PLCM document) permettra une harmonisation plus efficace dans les régions ICH.
Quant aux commentaires des industriels et des autres associations compilés par l’EMA, ils convergent globalement sur des parties de texte communes. Le commentaire général commun concerne l’harmonisation avec le cadre réglementaire « régional ». L’importance de retravailler sur la robustesse du Système de Management de la Qualité (Quality Management System) est souligné par tous.
5. Conclusions et perspectives
L’année 2019 a été consacrée à la revue des commentaires transmis par l’industrie et les associations. Deux sessions de travail du Groupe d’Experts ICH (Expert Working Group – EWG) ont permis l’adoption du texte final le 20 Novembre 2019. L’industrie découvre actuellement la version finale du guide et les choix qui ont été faits par l’ICH dans l’implémentation des commentaires. Nul doute qu’un travail important d’harmonisation est à prévoir avant une implémentation effective et efficiente du guide. Celle-ci demandera aux industriels et autorités réglementaires de collaborer pour décliner opérationnellement les concepts et s’aligner sur leur interprétation. La FDA a déjà initié cette logique d’échange, en lançant un programme pilote en 2019, dans lequel les laboratoires soumettant un dossier à évaluer ont pu proposer des Established Conditions et bénéficier d’une revue avec la FDA.
Les échanges réalisés lors des quelques mois de travaux du GIC A3P ont amené aux réflexions que les démarches QbD, basées sur la connaissance scientifique et le risque, vont permettre les démultiplications de gains financiers attendus, en autorisant que des Changements historiquement sous approbation soient implémentés sous la supervision du QMS. Cette vision, qui promeut l’innovation et l’amélioration continue, impose donc un QMS extrêmement robuste, sur l’ensemble de la chaîne de fabrication du médicament, et un Management des connaissances (“Knowledge Management”) opérationnel afin de valoriser toute la compréhension Produit / Procédé acquise en développement et complétée tout au long du cycle de vie du Produit.
Partager l’article


Johanne Piriou – AKTEHOM
Ingénieure en Biochimie/Biotechnologies (INSA Lyon), Johanne Piriou est consultante en maîtrise Produit/Procédés Pharmaceutiques chez AKTEHOM depuis 13 ans.
Johanne a mené des projets de mise en service, qualification, validation et démarrage d’unités de production (Produits finis stériles ou Drug Substance destinés à des produits finis stériles).
Au travers de ces projets de démarrage d’installation et en accompagnant des plans d’amélioration continue et de remédiation, Johanne s’est spécialisée dans la maîtrise de la contamination microbiologique et l’assurance de stérilité. Les domaines d’expertise de Johanne se complètent par des sujets tels que la mise en œuvre opérationnelle de l’approche QbD, la définition de Control Strategy et leur implémentation, ainsi que la Validation de procédés.
Aujourd’hui, Johanne pilote l’expertise Control Strategy, QbD, et Product Lifecycle Management au sein du cabinet de conseil AKTEHOM.
Bibliographie
(1) ICH Q12 – Technical and regulatory considerations for pharmaceutical product lifecycle management – Step 2 – Novembre 2017.
(2) Overview of comments received on ICH guideline Q12 on technical and regulatory considerations for pharmaceutical product lifecycle management (EMA/CHMP/ICH/804273/2017)
(3) Commission Regulation 1234/2008
(4) WHO/OMS – Guidelines on procedures and data requirements for changes to approved biotherapeutic products – WHO/PAC for BTPs_DRAFT/2016.
(5) Guidance for Industry – Changes to an Approved NDA or ANDA (2004)
(6) Guidance for Industry – CMC Postapproval Manufacturing Changes To Be Documented in Annual Reports (2014)
(7) FDA – GFI – Established Conditions: Reportable CMC Changes for Approved Drug and Biologic Products – Draft 2015.
Glossaire
AMM : Autorisation de Mise sur le Marché
CMC : Chemistry, Manufacturing, Control
CTD : Common Technical Document
EC : Established Condition
EMA : European Medicines Agency
EWG : Expert Working Group
GIC : Groupement d’Intérêt Commun
ICH : International Conference of Harmonization
KPP : Key Process Parameter
PACMP : Post Approval Change Management Protocol
PLCM : Product Life Cycle Management
PQS : Pharmaceutical Quality System
QbD : Quality by Design
QMS : Quality Management System
