


Sommaire
- Considerations for Validating Aseptic Manufacturing Processes.
- Simulation des procédés de fabrication aseptique de suspensions de nanoparticules ou microsphères : Challenges & facteurs de succès au travers d’un retour d’expérience.
- L’isolateur jetable pour le remplissage aseptique, innovation ou hérésie ?
- A3P/AFI Survey on Sampling & Testing Practices for In-Process Pre-Filtration Bioburden for Sterile Products: Presentation of the Results & Critical Discussion.
- Projet de norme internationale amené à être amendé PR ISO 11737-3 : Stérilisation des produits de santé - Méthodes microbiologiques - Partie 3 : essai des endotoxines bactériennes.
- Real-time detection of CFU growth with the ScanStation® smart incubator expedites the environmental monitoring process.
- MTP: What It Is, Why It Matters. Module type package standard is a major step forward for plug-and-produce manufacturing.
- How Pemflow / D.O.C. manage Extractables and Leachables Qualification of the in-process materials and primary packaging.
A3P/AFI Survey on Sampling & Testing Practices for In-Process Pre-Filtration Bioburden for Sterile Products: Presentation of the Results & Critical Discussion.
For sterile products and especially for parenteral products sterilized by filtration, the in-process pre-filtration bioburden test results are considered as key data for the assessment of the overall contamination control strategy performance.

This is clearly stated in the Introduction of the USP chapter <1229.3> “Monitoring of Bioburden”: “Monitoring of in-process bioburden of pharmaceutical components and products is an essential element of the overall contamination-control program for appropriate sterilisation process control. Bioburden monitoring should be designed for the recovery of a broad range of microorganisms that are likely to be present in the material being processed. Sterilisation processes are implemented in order to eliminate bioburden in materials and the products, ensuring both adequate process control and end-user safety. Bioburden is a potential risk to the patient not only because the sterilisation process might not be completely effective, but also post-processing because of the possible presence of residual materials such as allergens, endotoxins, and exotoxins. It may also have adverse impact on product quality and stability. Therefore, although bioburden may be confidently killed by destructive sterilisation processes or removed by retentive processes (filtration), […] its proliferation before sterilisation should be avoided. Process controls and cleaning, sanitization, and disinfection programs provide active means for the control of bioburden population and support the sampling, enumeration, and characterization of bioburden necessary to assure that sterilisation processes are effective”.
Key current regulatory expectations related to the in-process pre-filtration bioburden sampling and testing strategy for sterile products can be found in:
- The current version of the EU GMP Annex 1 Manufacture of Sterile Medicinal Products at point 80 “The bioburden should be monitored before sterilisation. There should be working limits on contamination immediately before sterilisation, which are related to the efficiency of the method to be used. Bioburden assay should be performed on each batch” […] “and considered as an in-process test.” ”Where appropriate the level of endotoxins should be monitored”.
- The EMA Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container (March 2019) at point 4.1.5 “For routine commercial manufacturing, bioburden testing should be performed on the bulk solution immediately before sterile filtration. In most situations, a limit of NMT 10 CFU/100 ml (TAMC) would be acceptable for bioburden testing. If a pre-filter is added as a precaution only and not because the unfiltered bulk solution has a higher bioburden, this limit is applicable also before the pre-filter and is strongly recommended from a GMP point of view”. “Bioburden should be tested in a bulk sample of 100 ml in order to ensure the sensitivity of the method. Other testing regimes to control bioburden at the defined level should be justified”.
- The FDA guidance for sterile products produced by aseptic techniques in section X.C. Prefiltration Bioburden “Manufacturing process controls should be designed to minimize the bioburden in the unfiltered product. In addition to increasing the challenge to the sterilizing filter, bioburden can contribute impurities (e.g., endotoxin) to, and lead to degradation of, the drug product. A prefiltration bioburden limit should be established”.
The EU GMP Annex 1 revision draft from February 2020 contains some new or modified requirements which underline an increasing emphasis put on this in-process test:
- Point 8.94 “Bioburden samples should be taken from the bulk product and immediately prior to the final sterile filtration. Systems for taking samples should be designed so as not to introduce contamination”.
- Point 10.3 “The bioburden assay should be performed on each batch for both aseptically filled product and terminally sterilized products and the results considered as part of the final batch review. There should be defined limits for bioburden immediately before the sterilizing filter or the terminal sterilization process, which are related to the efficiency of the method to be used. Samples should be taken to be representative of the worst case scenario (e.g. at the end of hold time)”.
- Point 10.4 “A pre-sterilization bioburden monitoring program for the product and components should be developed to support parametric release. The bioburden should be performed for each batch. The sampling locations of filled units before sterilization should be based on a worst case scenario and be representative of the batch. Any organisms found during bioburden testing should be identified and their impact on the effectiveness of the sterilizing process determined. Where appropriate, the level of pyrogen (endotoxins) should be monitored”.
This situation, with on one side, a growing emphasis on the in-process pre-filtration bioburden test for sterile products, and with on the other side, the current limited guidance on how to precisely perform the sampling, testing and how to manage the testing results, was at the origin of the survey on the current industry practices for this In- Process Bioburden test for sterile products launched by the A3P and the AFI in Q3 of 2021.
The objectives of the survey were:
- To collect current industry practices and trends ;
- To capture the differences in interpretation of the pre- filtration bioburden management expectations ;
- To assess any potential need for further clarification in current regulatory and pharmacopeial expectations for Parenteral Products in-Process Bioburden sampling, testing and results management.
The present article is divided in 2 parts.
The first part provides the main outcomes of that survey and opens the discussion around some concerns and clarification needs raised by the participants to ensure in the future more robust control strategies and to facilitate aligned interpretation between industry and regulatory authorities.
The second part of the article includes the inputs from recognized microbiological methods specialists who shared their opinion and perspective on the current status of the in-process pre-filtration expectations for sterile products:
- Thierry Bonnevay, Global Microbiology Analytical Expert, Sanofi Vaccines / Member of the European Pharmacopeia Expert Group N°1 (Microbiology).
- Radhakrishna Tirumalai, Senior Principal Scientist, Merck Research Laboratories Center for Excellence in Microbiology / Former Senior Principal Scientist and Liaison to the USP General Chapters-Microbiology Expert Committee at USP Convention
- Di Morris, PHSS Vice Chairperson / Former Medicines Inspector of the UK Department of Health.
1. Survey Results
1.1 Key Figures
The survey included 20 questions posted during Q3/Q4 2021 by the A3P and AFI. For the purpose of the following results analysis, the answers to several questions or sub-questions were grouped.
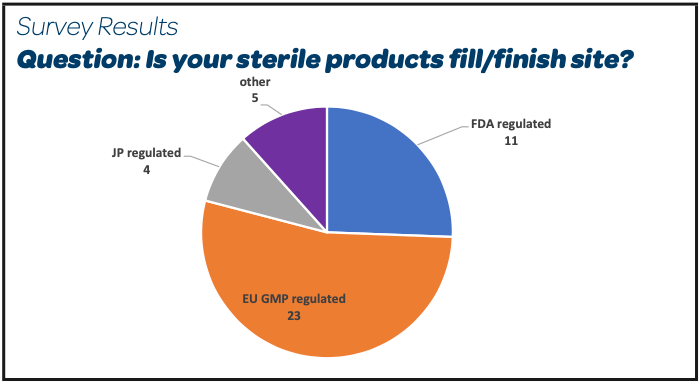
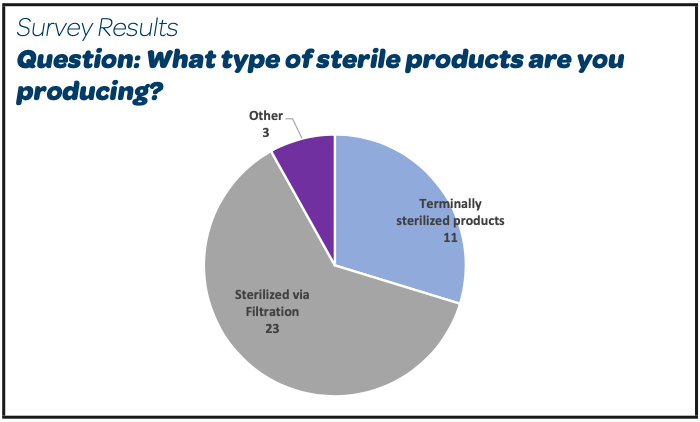
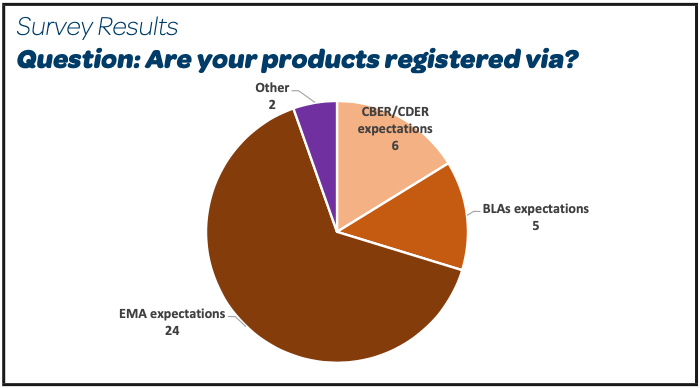
There were 26 participants, mainly EU or FDA GMP regulated sites, producing parenteral products sterilized by filtration and registered as per EMA expectations (Other: Global Markets, Russia, Swiss Medic).
 |  |  |
1.2 Registration practices
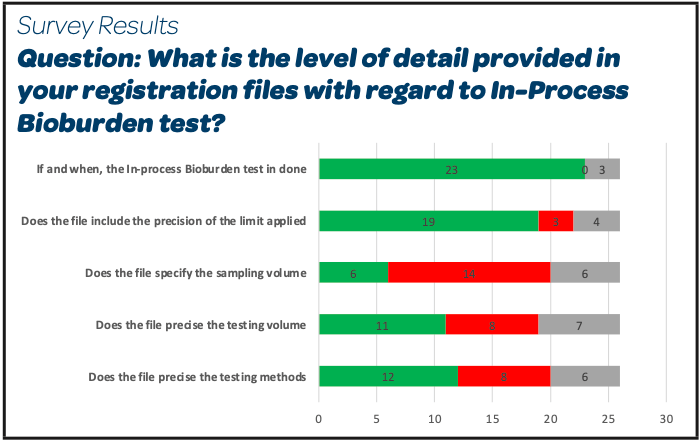 |
- A large majority of the participants register the in-process bioburden test, its associated limit and the location of the sampling
- Almost 50 % precise the testing method and volume. Sampling volume is less frequently included (6/26)
- It is noticed that 4 companies participating to the survey have answered yes to all these questions with the precision in the comments sections that this level of precision was directly expected by the registration dossier assessors.
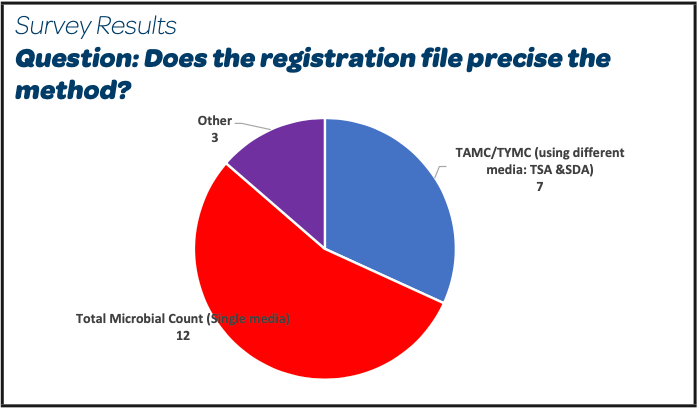 |
- More than 50% of the participants register Total Microbial Count (TMC) using Trypticase Soy Agar media (TSA) as the method test for in-process bioburden
- 7/26 declare Total Aerobic Microbial Count (TAMC with TSA media) and Total Yeasts and Molds Count (TYMC using Sabouraud Dextrose Agar SDA); among them 3 participants add a selective growth agent into the SDA media.
- 2 participants have also registered TMC or TAMC and TYMC depending on the registration period.
- 3 participants did not answer this question.
1.3 Sampling Practices
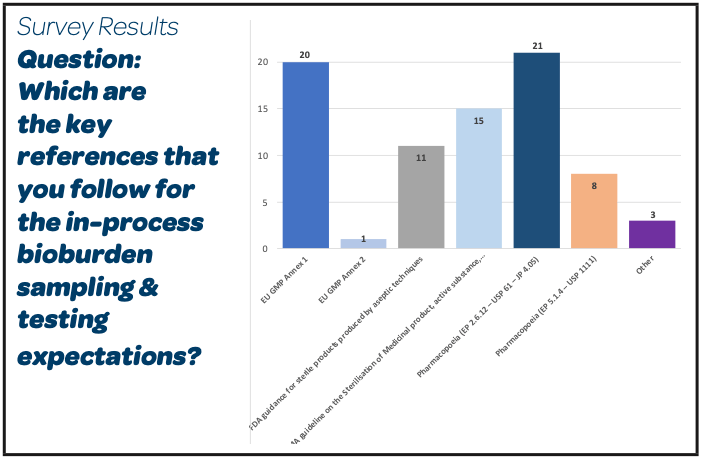 |
- With regard to sampling and testing requirements for In-Process Pre-Filtration Bioburden test of Sterile Products, a large majority of participants (21/26) do apply the Pharmacopeial expectations for the Microbial Numeration test to use for the release of Non-Sterile Products (EP 2.2.12 – USP 61 – JP 4.05)
- 20/26 use also the EU GMP Annex 1 and 15/26 the EMA guideline on the Sterilisation of medicinal product, active substance, excipient and primary container. Most of them use these references in addition to the pharmacopeias.
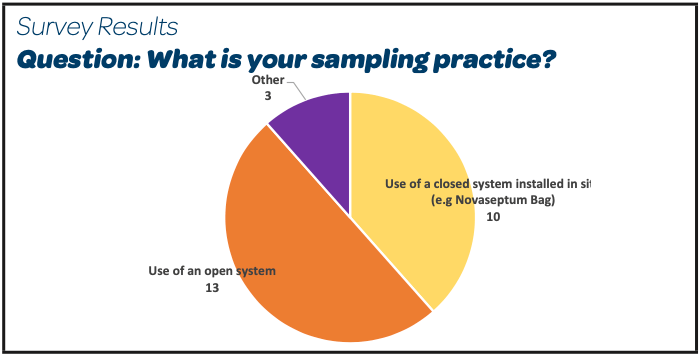 | 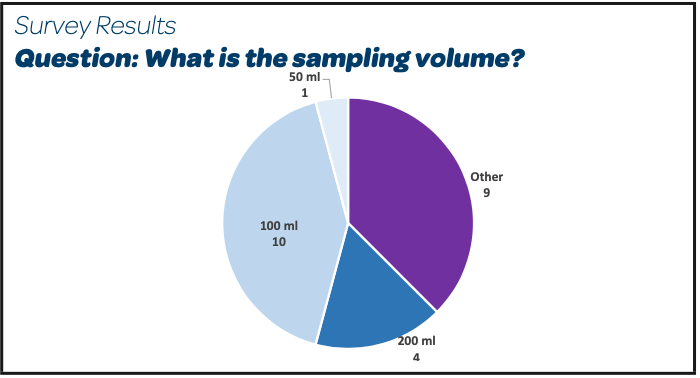 |
- 16/26 perform a product bioburden sample before any filtration step
- 13/26 continue to use open systems to collect that sample
- We can observe that several different sampling volumes are used; a detailed analysis of individuals answers shows that 15/26 have a sampling volume of 100 mL or a larger volume (200 – 250 – 300 mL)
- In the case of small bioproducts production batches, the trend is to sample less than 100 mL
- One answer included as a comment that 100 mL is the sampling volume before the sterilizing filter, but a lower volume (10 mL or even 1 mL) is sampled before the bioburden reduction filter.
1.4 QC Lab testing practices
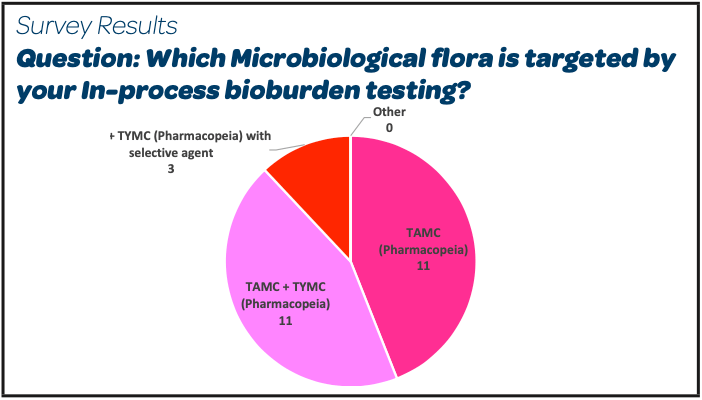 | 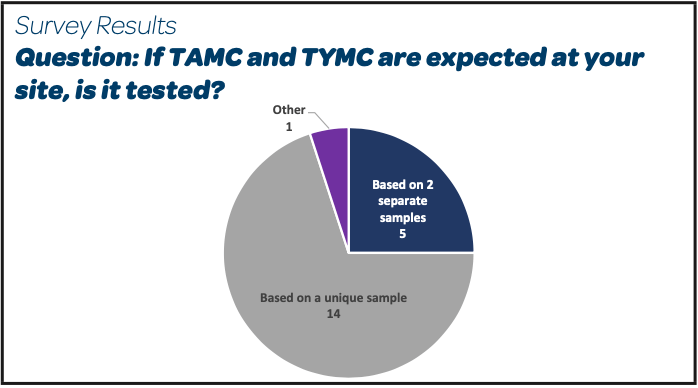 |  |
- The microbiological flora targeted is TAMC and TYMC in a short majority (14/26). A detailed analysis of the individual answers shows that among these 14 answers, the TAMC and TYMC tests are done based on a unique sample of product around 75 % of the cases and done on 2 separate samples in the other 25 %.
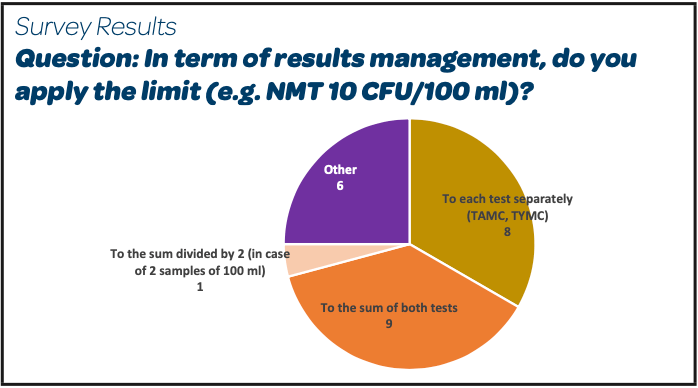
- There is no real trend observed in term of results management as approximately 50 % of the respondents (8/18 for that question) do apply the limit of NMT 10 CFU/100 ml to each test method separately and approximately 50 % (9/18) on the sum of both test methods.
- 1/18 respondents makes the sum of the results of TAMC + TYMC divided by 2 and applies the limit of NMT 10 CFU/100 mL to the obtained result.
- The rationale provided by the participants who are targeting “TAMC only” is that the EMA guideline for sterilisation of the medicinal product, active substance, excipient and primary container sterilisation provides this limit of NMT 10/100 mL for TAMC “acceptable in most situations”.
- Among the participants who are targeting TAMC + TYMC, 3 have added a selective agent to avoid that bacteria such as Gram positive spore formers (that can be recovered on both media) are counted twice and so the limit of NMT 10/100 mL applied to the sum of the 2 results may be seen as more relevant.
1.5 QA/QC practices
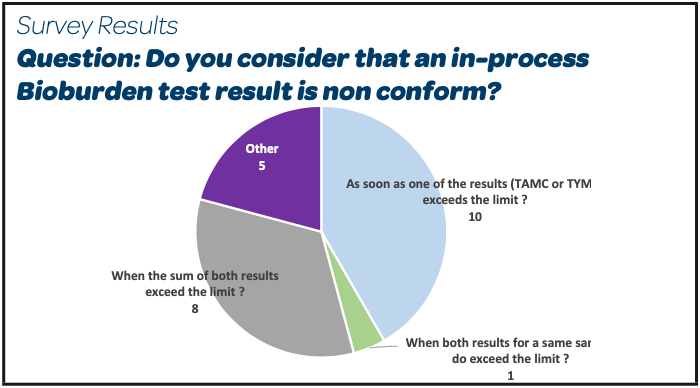 | 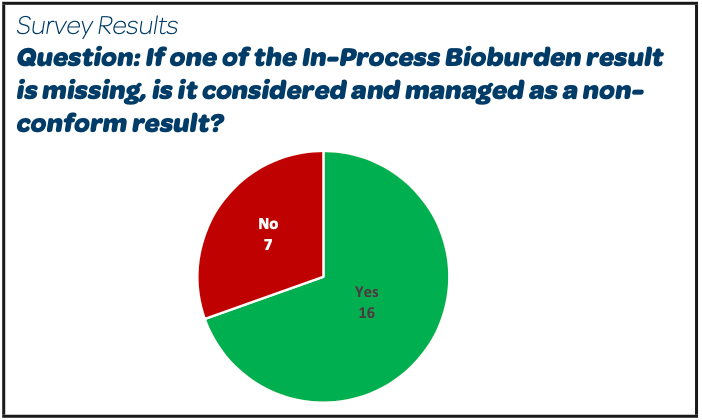 | 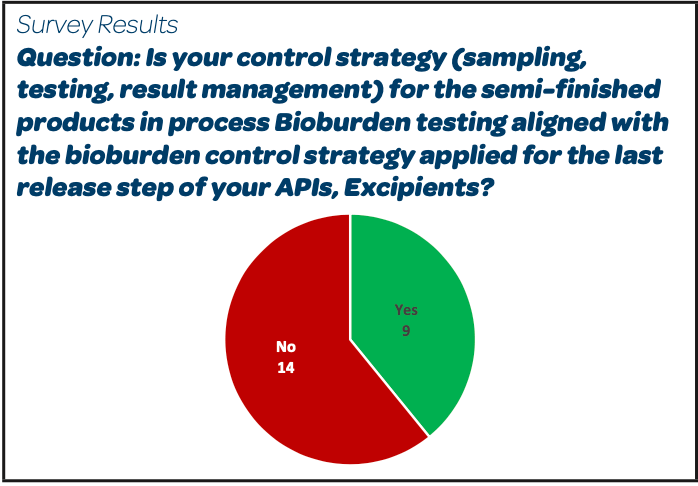 |
- 10/24 respondents do consider that the in-process bioburden test is non-conform as soon as one of the results (TAMC or TYMC) exceeds the limit. 8/24 only when the sum of both exceeds the limit.
- The majority (16/23) handle a missed bioburden sample as they would for a non-conform result (decision on the impacted batch based on the investigation, the additional process and other product testing data, microbiological test results for APIs and excipients, filter validation data, absence of observed trend for the in-process bioburden test results, etc).
- 14/25 respondents do not have the same strategy for bioburden sampling, testing and results management between the last release step of their API and for the in-process bioburden of the semi-finished products using the API.
1.6 Registration and Regulatory Inspections Trends
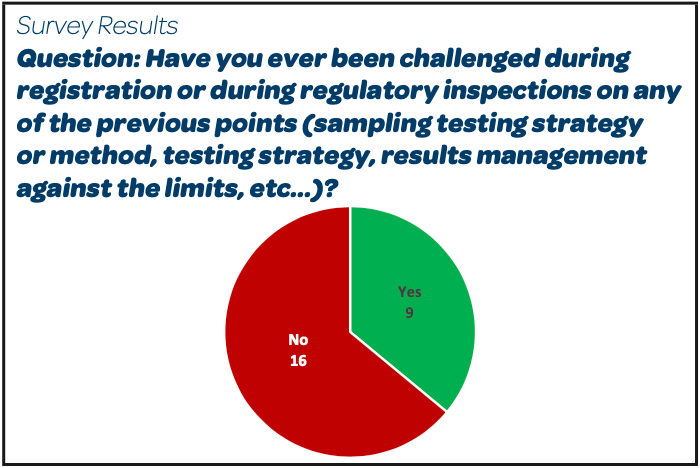 |
Reported Challenges / Observations:
- “No bioburden sampled before first filtration step”
- “Use of an open system for the sampling”
- “Non adequate specification used for the bioburden performed on the compounding tank before the clarification filtration (= before the storage and before sterile filtration)”
- “Bioburden sampling is done prior to the filling, only at the beginning and not at the end of final transfer”
- “EU Authorities: sampling volume non conform to 100 mL indicated in the guideline for sterilisation of medicinal products”
- “Absence of rationale to support the fact that we do not test 100 mL but 10 mL on TSA and 10 mL on SDA”
- “In-Process Pre-filtration Bioburden limit of 10 CFU/100 mL not applied to the sum of TAMC+TYMC result as done for raw materials”
1.7 Key potential clarification needs
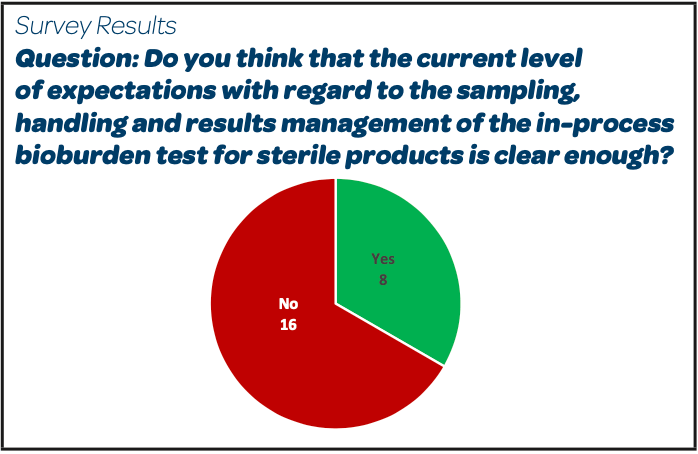 |
Abstract of collected Comments and Expressed Needs:
- “Some margin must be left to companies depending on the product context and risk profile”.
- “Missing information about the expected rationale if volume differs from 100mL. Same for the calculation method for combining TAMC and TYMC”.
- “No regulation clearly describes the bioburden before a sterile filtration or for contamination control strategy. Too much confusion with microbial count for non-sterile products”.
- “We should clarify the result management when we test on TSA and SDA and the need to perform the sum”.
- “Need to understand the management of the results”.
- “Clarification on the method to be used for the expected result of bioburden of NMT 10 CFU/ 100 mL for the pre-sterilisation bioburden and a guideline for other filtration steps reference limits will be the best to avoid the multiple interpretations and strategies adopted by the different companies and in some cases also different sites of the same company”.
- “Unfortunately, due to categorization of IPC, pharmacopeias do not want to describe the method to be applied in detail. Some Inspectors make the confusion with microbiological quality of final non-sterile products”.
1.8 Discussion & Conclusion
The presented survey results on the current practices on in-process pre-filtration bioburden for sterile products show a real variability of in sampling, testing, results management and a certain level of confusion and misalignment on the interpretation of applicable regulatory or pharmacopeial expectations at industry and at inspection levels.
The direct application of release testing monographies for non-sterile products to in-process testing for sterile products without considering the overall bioburden control program in a sterile product facility for a specific production and sterilisation process, so as the product specificities seems inadequate.
The USP chapter <1229.3> Monitoring of Bioburden provides the following important knowledge elements.
a. On semi-finished product specificities
“Many products are inherently antimicrobial, and some formulations contain antimicrobial preservatives, both of which can limit bioburden recovery. Products that are outside of the pH range of approximately 4-9 are strongly hypertonic or strongly hypotonic, or have low water activity, may reduce the level of recoverable microorganisms”
b. On product bioburden control strategy
A typical bioburden-control program includes review and analysis of potential sources of contamination as well as sound process design and preventive and monitoring measures. The microbiological contamination-control program should be developed to identify and control bioburden and to asses product risk based on a formal assessment of risk modalities. The bioburden risk assessment should result in the establishment of critical control points and should include consideration of the following elements :
- Microbiological attributes of materials before sterilization and the manufacturing process used for the materials (if applicable)
- Inherent antimicrobial properties oh the materials
- Time limits for process execution
- Water activity of the material
- Environmental conditions within the facility
- Equipment design and cleaning
- Sanitization, decontamination, and other active microbial control processes (such as pre filtration, temperature, pH, osmolarity, etc.)
Conrolling the bioburden of materials and products to be sterilized will ensure to the levels required by the sterilization process validation. Additionally, controlling the biobruden levels of the items to be sterilized assures that residuals (e.g., allergens endotoxins, and exotoxins) form that population will also be controlled. This is important because direct detection of the materials is challenging.
c. On sampling and testing considerations
- “Bioburden evaluation should focus on micro-organisms that represent the greater concern on the sterilisation process. Total count methods should consider the properties of both the product under evaluation and the characteristics of the process”. “Bioburden of greatest concern for a sterilizing filtration are Pseudomonas, Brevundimonas, Ralstonia and Mycoplasma”
- “The methods listed in Microbiological Examination of NonSterile Products: Microbial Enumeration Tests <61> may be appropriate for bioburden evaluation, although they may require modification”.
In conclusion, based on the survey results, trends and concerns/ difficulties raised by the participants but also by taking into account the listed knowledge elements, is appears that there is an opportunity for adequate experts to develop additional guidance that would help the industries and the regulators to align on a best practice in term of sampling, contamination screening, testing methods and test results management for sterile products in process pre-filtration bioburden.
2. Experts view
This second part of the article consists in the presentation of the interview answers from 3 experts to a questionnaire focused on the 3 main concerns/difficulties raised in the first part.
Our expert panel includes:
- Thierry Bonnevay, Global Microbiology Analytical Expert from Sanofi Vaccines, Member of the European Pharmacopoeia Microbiological Methods Experts Group N°1 Microbiology -> answers in green.
- Radhakrishna Tirumalai, Senior Principal Scientist, Merck Research Laboratories Center for Excellence in Microbiology Former Senior Principal Scientist and Liaison to the USP General Chapters Microbiology Expert Committee at USP Convention -> answers in blue.
- Di Morris, PHSS Vice Chairperson, former Medicines Inspector of the UK Department of Health -> answers in purple.
Topic 1: Applicable expectations to in-process pre-filtration bioburden for parenteral products
Question 1: Which is the most relevant regulatory expectation or guidance to follow to ensure a compliant and relevant strategy for in- process pre-filtration bioburden strategy for parenteral products?
- “The EMA Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container (March 2019)” because it’s the only regulatory document that precises the volume to be tested (100 mL), the specification to be applied (NMT 10 CFU / 100 mL) and the method to be used (TAMC, so only Soybean soja agar media incubated at 30-35°C during 3 to 5 days)” (TB)
- “I am not aware if any relevant regulatory guidance exists on this topic that can be followed without modification” (RT)
- “The EMA Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container (from March 2019 = EMA/CHMP/CVMP/QWP/850374/2015 that came into force 1 October 2019)
This guideline replaces the document Decision trees for the selection of sterilisation methods (CPMP/QWP/054/98), which is an annex to the note for guidance on development pharmaceutics (CPMP/QWP/155/96) and the document Decision trees for the selection of sterilisation methods (EMEA/CVMP/065/99) which is an annex to the note for guidance: Development pharmaceutics for veterinary medicinal products (EMEA/CVMP/315/98).
However, it missed the reference to CPMP/QWP/486/95 – but it contains the same information
- Legal Status – This guideline should be read in conjunction with Directive 2001/83/EC on the community code relating to medicinal products for human use, Directive 2001/82/EC on medicinal products for veterinary use as amended and also the current European Pharmacopeia. It is legally binding and should be followed.
- In addition, this guideline should be read in conjunction with all other relevant directives and regulations, and all relevant Commission, (V)ICH and CXMP guidelines, Q&A documents and other documents as linked to or published on the EMA website (www.ema.europa.eu)
The product bioburden is a requirement of the dossier and not the Pharmacopoeia or Annex 1 with regard to the pre filtration bioburden limit. It has a link to GMP hence inspectorates are checking bioburden results.” (DM)
Question 2: What do you think about the extension of some microbial numeration requirements for final release of non-sterile products to in- process pre-filtration bioburden testing of parenteral products?
- “These are just two different things: IPC bioburden before filtration is a quantification and identification of the bioburden done to know what is immediately going on the filter for assessment of the efficacy of sterile filtration (Microbial retention). For final non-sterile product, it reflects the final non-sterile product microbiological quality that results from the entire processing steps (storage and shelf life included).”
- “Yes, if so, only TAMC as an approximation, with an Endotoxin specification, as well”. (RT)
- “That assessment has to be included in the Contamination Control Strategy – for a sterile product the expectations for the enumeration are clear – NMT10cfu/10mL for the solution prior to the final filtration. The regulatory and inspectorate position is that the bioburden has to meet these limits using the test method from the Ph.Eur. and that it is the solution before the sterilising filter – ie the one as close as possible to the fill line as per Annex 1 that has to comply with NMT10cfu/100mL The regulation is clear – it specifies the value and when it should be taken. It states the test to be used in the Ph.Eur. It states that it has to be part of the description in the dossier and it has its legal basis in the directives.” (DM)
Question 3: How far is this relevant? (e.g systematic search of yeast and molds on an additional media that is not necessarily selective enough, make the sum of the TAMC and TYMC results generated on 2 separate testing samples of 100 mL and applying the acceptance criteria of NMT 10 CFU/100 mL on that sum, etc…)
- “IPC for Product bioburden before filtration has the same specification as Water for Injection. Testing 100 mL for high production volumes for TAMC only is sufficient based on the fact that the product sampling in performed in a filling area Grade A/B, with no molds at all or less than 0,1% of mold recovery in the environment, that TAMC method enables the recovery of a large variety of molds et molds are the less challenging micro-organisms to retain by filtration (x10 at least bigger than bacteria). So only TAMC, with 100 mL product analyzed by membrane filtration, one final result and specification < 10 CFU / 100 mL is appropriate. Don’t forget that this test is not only a quantitative assay but also a qualitative assay, any CFU recover by the method should be identified to the species level even if you are compliant to the specification (less or egal to 10 CFU). In the case of bulk size less than 5 liters, you could sample about 1% of the total production volume.” (TB)
- “An additional general media would be the best solution.” (RT)
- “EP 2.6.12 allows for a single media which has yeast and mould growth promotion. I do not understand why organisations think they need to do a separate Yeasts and Moulds (unless mandated by a competent authority) – if that was the case, we would still be using both media all the time in our EM programmes. I think the bigger picture is why are folks thinking they need to use SDA – both the regulation and E.P. 2.6.12 refer to TMAC and not Yeasts and Moulds knowing that this is the test to be applied with as I said the correct growth promotion. We know that the media we use is already limited and does not recover the vast majority of organisms and will not recover sub- lethally challenged organisms hence the importance of controlling bioburden from the start of the process and is the main impact of a Contamination Control Strategy”. (DM)
Topic 2: Control strategy details (flora targeted, media to use, sampling & testing volume, limit)
Question 1: Based on the survey questionnaire results, we observe that different control strategies are applied that are not always based on strong rationales. How can we explain this variability in the current interpretation of the applicable expectations at industry and regulatory inspection level?
- “Because there is clearly a confusion at industry and inspectors levels between regulations that are clear for release test of final non sterile product and an IPC test for immediate bioburden to challenge the filter used for sterilisation purpose”. (TB)
- “Lack of clear guidance or understanding and misapplication of methods including sampling”. (RT)
- “It goes back to people just using the PhEur and not reading the regulation that goes with it for pre filtration bioburden. It is clear that not everyone is aware of the legal duties and expectations and that not all assessors are working to the same requirements” (DM)
Question 2: In the context of the new EU GMP Annex 1 revision that promotes the use and documentation of risk-based strategies, do you think that an appropriate process/product risk assessment as the basis of the strategy would be better accepted in the future by the inspection authorities?
- “Yes. Explain and justify why only TAMC with 100 mL and specification < 10 CFU/100mL are sufficient”. (TB)
- “Yes, I agree with a risk-based approach”. (RT)
- “No, because risk assessments performed by most of the industry is to provide a justification for not doing something rather than the full impact to the patient; a risk assessment should be to meet the regulation but in a different way – the regulation already allows for a different process with appropriate justification”. (DM)
Question 3: If yes, which aspects should be included in such a risk assessment?
- “The challenge used for the sterilizing filter, the very low mold recovery in the production areas, the fact that almost all the main encountered molds can growth well on TAMC test media. In addition, most of us do not use the Sabouraud as part of our environmental monitoring program for the same reasons”. (TB)
- “Total counts, type (gram negative, for example) and size”. (RT)
Topic 3: Possible clarifications at pharmacopeial level? Or in any other regulatory reference?
Question 1: Is there any initiative in-progress at pharmacopeial level to clarify the expectations regarding in-process pre-filtration bioburden Bioburden for parenteral products? at other regulatory institution level?
- “Not for Ph Eur, and I’m not aware about such clarification for other Pharmacopeias. If so, would be more a guideline than a compendial assay”. (TB)
- “Yes, at USP”. (RT)
- “It is clear already – organisations just need to understand the basis for the regulation”. (DM)
Question 2: If yes, please share the scope and status of current actions?
- “Either a new chapter or revisions to existing USP chapter <1229.3> + an article clearly describing the intent and limitations of USP <61> is planned. A chapter on Definitions (lexicon) is also being proposed. The target for these changes is a publication at the Pharmacopeial Forum by mid-2023”. (RT)
Question 3: What would you recommend to ensure a stronger understanding of the real expectation for that test and a better alignment in term of interpretation of the expectations between industries and authorities?
- “Scientific articles (PDA, biological, USP, A3P La Vague, etc..), future guideline in pharmacopeias, oral presentations during conferences advocate and explain to microbiology QC head as well as to inspectors the current regulation to avoid wrong extension of non- sterile product release test expectations to an In Process Control test before the sterile filtration for parenteral products”. (TB)
- “We have to understand correctly the definition of Bioburden and apply/develop a method that will address bioburden. We know that bioburden is not merely sum of TAMC and TYMC”. (RT)
- “That QA and regulatory compliance ensure they understand the legal basis for the requirements and top follow them, EMA has not helped by not ensuring consistency in the history between the documents as they have been updated but regulatory departments should be following the changes. The expectation for the test is to ensure there is a low bioburden in the solution before it reaches the final filtration. A low bioburden should reduce the risks on endotoxin and exotoxins contaminating the final product and passing the LAL test – which has its own limitations and is a key part of a products sterility assurance and patient safety.” (DM)
Conclusion for the experts’ views part:
Each of our 3 interviewed specialists has provided his/her own perspective and opinion based on expertise background, experience, sensibility and regional interpretation (each expert comes from another country France, USA or UK). The legal fundamentals were reminded and summarized so as the way we must apply the texts. Multiple rationale elements have been provided that could be further re-used to develop additional risk-based guidance for sampling, testing methods and testing results management.
Work is in-progress at USP level that hopefully will bring clarity in how far the application of pharmacopeial expectations for non-sterile products release test can be extended to the in-process bioburden test for sterile products.
Partager l’article

Références
- Current EU GMP Annex 1 Manufacture of Sterile Medicinal Products (2008)
- EU GMP draft revision Annex 1 Manufacture of Sterile Medicinal (2020)
- FDA guidance for sterile products produced by aseptic techniques (2004)
- EMA Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container (2019)
- USP chapter <1229.3> Monitoring of Bioburden
- Harmonized Pharmacopeia (EP 2.6.12 – USP 61 – JP 4.05) Microbial Enumeration Test for NonSterile Products
- USP chapter <1229.4> Sterilizing Filtration of Liquids
- Hoenen I, Short overview of the A3P/AFI group Survey Results on In-Process Pre- Filtration Bioburden Testing for Sterile Products, A3P Microbiological Form, Lyon, June 2022.

