


Sommaire
- Considerations for Validating Aseptic Manufacturing Processes.
- Simulation des procédés de fabrication aseptique de suspensions de nanoparticules ou microsphères : Challenges & facteurs de succès au travers d’un retour d’expérience.
- L’isolateur jetable pour le remplissage aseptique, innovation ou hérésie ?
- A3P/AFI Survey on Sampling & Testing Practices for In-Process Pre-Filtration Bioburden for Sterile Products: Presentation of the Results & Critical Discussion.
- Projet de norme internationale amené à être amendé PR ISO 11737-3 : Stérilisation des produits de santé - Méthodes microbiologiques - Partie 3 : essai des endotoxines bactériennes.
- Real-time detection of CFU growth with the ScanStation® smart incubator expedites the environmental monitoring process.
- MTP: What It Is, Why It Matters. Module type package standard is a major step forward for plug-and-produce manufacturing.
- How Pemflow / D.O.C. manage Extractables and Leachables Qualification of the in-process materials and primary packaging.
Projet de norme int. amené à être amendé PR ISO 11737-3. Stérilisation des produits de santé - Méthodes microbiologiques - Partie 3 : essai des endotoxines bactériennes.
Les Essais des Endotoxines Bactériennes (EEB) sur les dispositifs médicaux (DM) et produits de santé sont conduits actuellement selon des méthodes décrites dans les référentiels suivants :
- PE 2.6.14 : Essais des endotoxines bactériennes – USP <85> : BACTERIAL ENDOTOXINS TEST
- USP <161> MEDICAL DEVICES: BACTERIAL ENDOTOXIN AND PYROGEN TESTS
- L’ANSI/AAMI ST72 : 2019 (Bacterial endotoxins- Test methods, routine monitoring, and alternatives to batch testing)

Les méthodes des Pharmacopées européennes et américaines concernent davantage les produits pharmaceutiques et ne sont pas spécifiques aux DM, à l’exception de l’USP <161>.
L’ANSI/AAMI ST72 étant une norme américaine, ce projet de norme internationale est donc indispensable pour combler le vide normatif au niveau international. Ce dernier a débuté en 2018 et est actuellement au stade DIS d’Enquête Publique (Résultats du dépouillement de l’enquête communiqués le 25/03/2022), la publication de la nouvelle norme devrait avoir lieu en 2023. Il s’est largement inspiré de l’ANSI/AAMI ST72 : 2019, même si plusieurs sections, tant dans la partie normative que dans la partie informative, ont été réorganisées, étendues ou modifiées.
À la suite des récents résultats du dépouillement de l’enquête publique montrant une opposition majoritaire à ce projet en raison du fait de l’exclusion de méthodes alternatives, ce dernier sera amené à être modifié. Dans l’attente de la version finale de la norme, l’objet de cet article est de mesurer l’impact qu’elle pourra avoir sur les EEB des fabricants de produits de santé et de DM en particulier, en détaillant les principales modifications des méthodologies engendrées versus celles des Pharmacopées (PE ou USP).
1. Objet / Domaine d’application
L’enjeu de cette nouvelle norme est de décrire les critères généraux à appliquer pour la détermination des endotoxines bactériennes présentes sur ou dans les produits de santé, les composants ou les matières premières en utilisant les méthodes d’EEB, à l’aide des réactifs de Lysat d’Amébocyte de Limule (LAL). Elle ne s’applique donc pas à l’évaluation des pyrogènes autres que les endotoxines bactériennes.
Les autres méthodologies de détection des endotoxines (Essais d’Activation des Monocytes ou MAT et le facteur C recombinant ou rFc), ne sont pas incluses. Les spécifications particulières de limites d’endotoxines ne sont pas non plus abordées dans cette norme.
Des recommandations sont formulées au sujet de la sélection des unités de produit, de la validation des
méthodes, de l’utilisation des techniques pour les essais de routine, de l’interprétation des résultats d’essai, des alternatives aux essais par lots et à l’évaluation des risques.
Les essais décrits dans cette norme portent notamment sur les produits devant être non pyrogènes en raison de leur usage prévu et/ou de la mention “non pyrogène” sur l’étiquette.
Les produits qui doivent être non pyrogènes ou qui sont étiquetés comme tels (Tableau 1) doivent faire l’objet d’une justification explicite au moyen d’une méthode EEB appropriée.

Cette justification doit comprendre au moins l’un des éléments suivants qui constituent les 2 types de plans d’échantillonnage :
- Essais sur le produit fini pour chaque lot (essais par lots)
- Alternative aux essais par lots.
2. Sélection des unités de produits et Plans d’échantillonnage des essais EEB
La sélection des unités de produits pour les essais EEB doit être fondée sur des critères définis dans le plan d’échantillonnage qui doit comporter les échantillons représentatifs des familles de produits établies sur la base d’une évaluation des produits, des procédés de fabrication, des composants et des matériaux.
Des recommandations pour définir ces groupes d’échantillonnage comme lot de production aux fins de l’essai des endotoxines (plusieurs types ou groupes de produits similaires présentant le même risque d’endotoxines) sont données en Annexe A de la norme.
Ce plan d’échantillonnage peut être de 2 types :
- Essais par lots
L’absence de pyrogène est confirmée par l’utilisation d’essais sur le produit fini.
Le lot peut être défini comme étant chaque lot de production ou un groupe d’échantillonnage autre que le lot de production sur la base d’une justification documentée ou d’une évaluation des risques.
Des recommandations sont données dans cette nouvelle norme pour déterminer le nombre d’échantillons nécessaires en fonction de la taille du lot (Tableau 2).
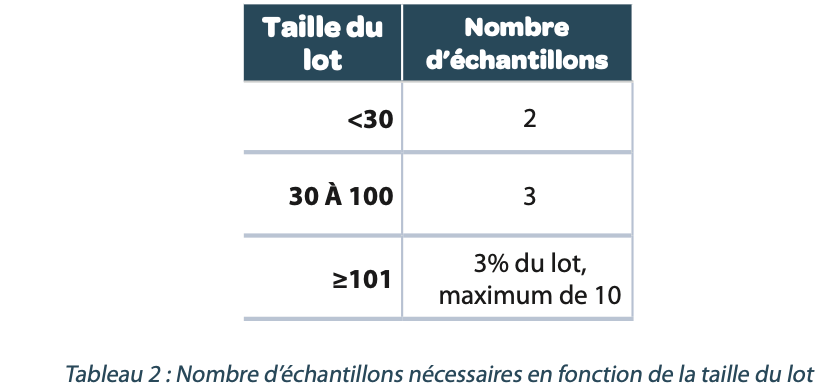
Dans la plupart des cas, chaque lot de produit doit être soumis à essai en utilisant un nombre approprié d’échantillons, ne dépassant pas 10, pris au hasard pour représenter la qualité du lot. Les plans d’échantillonnage alternatifs qui utilisent des échantillons de petite taille ou qui ne soumettent pas à essai chaque lot de produit doivent être clairement définis et étayés/justifiés par une évaluation des risques.
D’autres plans d’échantillonnage dérivés statistiquement, nécessitant parfois un plus grand nombre d’échantillons, peuvent être nécessaires à des fins de validation ou d’investigation.
Si les plans d’échantillonnage utilisés nécessitent la sélection de plus de 10 échantillons d’un lot, il convient de ne pas regrouper plus de 10 échantillons dans un essai.
- Alternatives aux essais par lots
Des alternatives aux essais par lots peuvent être utilisées s’il a été démontré que les procédés de fabrication, les matériaux et l’environnement de fabrication sont bien contrôlés.
Si des alternatives aux essais par lots sont utilisées, une évaluation des risques doit être effectuée afin d’évaluer les critères utilisés pour établir le plan d’échantillonnage : voir Paragraphe 7.
Les échantillons sélectionnés pour les essais doivent inclure tous les facteurs susceptibles d’avoir une incidence sur les niveaux d’endotoxines ou d’y contribuer.
3. Méthodes pour l’EEB
Il n’existe aucune différence de méthodologies avec celles décrites dans les Pharmacopées Européenne et Américaine PE 2.6.14, USP <85> et USP <161> :
Les trois techniques pour les EEB décrites dans la Norme sont les mêmes que celles des Pharmacopées précitées :
- Techniques par gélification : essai limite et méthodes de dosage
- Techniques en point final : colorimétrie en point final
- Techniques cinétiques : colorimétrie et turbidimétrie cinétiques.
Concernant les calculs de la limite d’endotoxines applicable pour la solution d’extrait et de la Dilution Maximale Significative (DMS), les formules de calcul sont les mêmes que celles décrites dans la Pharmacopée américaine USP <161>.
Les paramètres d’essais critiques, équipements / matériaux et réactifs recommandés sont également les mêmes que ceux décrits dans les Pharmacopées Européenne et Américaine.
4. Validation des méthodes pour l’EEB
Elle comprend la validation du produit et de la méthode d’essai, la préparation des échantillons ainsi que la qualification des réactifs et des analystes. Au moins un lot par produit ou famille de produits doit être utilisé afin de démontrer la validation du produit et de la méthode d’essai.
4.1 Validation du produit et de la méthode d’essai
Quelle que soit la technique utilisée (méthodes par gélification, cinétique ou en point final), les exigences pour la validation du produit/ de la méthode d’essai et le nombre de réplicats (ou d’exemplaires) restent les mêmes que celles des Pharmacopées européenne et Américaine.
Les seules différences apportées par la Norme par rapport à ces dernières ont trait :
- A la possibilité d’utiliser des Courbes d’étalonnage archivées (Tableau 3) pour la série de contrôle standard lors de la validation des méthodes d’essais pour les méthodes cinétiques et en point final, ce qui peut constituer un gain de temps appréciable et limiter les manipulations source d’erreur.Au calcul du recouvrement moyen de l’endotoxine ajoutée (PPC): “Calculer le recouvrement moyen en soustrayant la concentration moyenne en endotoxine dans la solution d’échantillon de la concentration moyenne en endotoxine dans le contrôle positif du produit et en divisant par la concentration en endotoxine connue” (alors qu’il est écrit dans les Pharmacopées : “Calculez le recouvrement moyen des endotoxines ajoutées en soustrayant la concentration moyenne en endotoxines de la solution seule de celle obtenue pour la solution contenant les endotoxines ajoutées”).
- Aux critères de validation de la méthode : Les 3 critères cités dans la Norme sont identiques à ceux des Pharmacopées excepté celui concernant le résultat obtenu pour le contrôle négatif (Solution D) : La Norme précise que “le temps de réaction moyen du contrôle négatif doit être supérieur au temps de réaction moyen du standard le plus faible de la série de contrôles” alors que les Pharmacopées demandent que le “résultat obtenu pour la solution D (témoin négatif ) ne soit pas supérieur à la limite spécifiée pour le blanc dans la description du lysat utilisé, ou est inférieur à la limite de détection de ce lysat.”
- A la possibilité d’utiliser des Courbes d’étalonnage archivées (Tableau 3) pour la série de contrôle standard lors de la validation des méthodes d’essais pour les méthodes cinétiques et en point final, ce qui peut constituer un gain de temps appréciable et limiter les manipulations source d’erreur.
L’Annexe B.15 apporte les précisions suivantes
Depuis la publication de l’ANSI/AAMI ST72 : 2019, des techniques et des équipements automatisés permettant de réduire les variations et les erreurs ont été développés et utilisés plus largement. Par exemple, un système automatisé de manipulation des liquides, l’utilisation de robots, etc. La plateforme mesure l’absorbance et compare la valeur observée avec une courbe d’étalonnage archivée.
Weber et al. (2014) ont rapporté que l’unité a réduit la manipulation des échantillons, le temps d’essai, les écarts et les investigations liées aux EEB.
4.2 Préparation des échantillons
Des recommandations sont apportées pour la préparation et l’extraction (ou non) des échantillons en fonction de leurs caractéristiques physiques (produits de santé solides, aqueux).
Des solutions pour pallier les interférences de l’échantillon (inhibition ou activation) sont également proposées : les extraits de l’échantillon peuvent être dilués sans dépasser la DMS et/ou traités par filtration, neutralisation, dialyse ou traitement thermique.
Ces traitements doivent être validés ou leur adéquation doit être démontrée sans perte d’endotoxines.
Toutes les manipulations d’échantillons doivent être spécifiées dans les données/rapports sur la validation de la méthode, et le même procédé doit être suivi pendant les essais de routine.
Contrairement à la Norme ISO 11737-1 pour laquelle une validation de l’efficacité de récupération de la biocharge est requise, la validation de l’efficacité de l’extraction pour les essais de détection d’endotoxines n’est non seulement pas demandée, mais elle n’est pas recommandée. En effet, le groupe de travail sur les méthodes microbiologiques du comité de stérilisation de l’American Association of Medical Instrumentation (AAMI) chargé en 2004 d’évaluer la pertinence de l’efficacité de la récupération des endotoxines pour l’extraction de produits de santé en vue d’essais sur les endotoxines, a montré que les limites d’endotoxines établies par la FDA et l’USP ont un facteur de sécurité adéquat basé sur une récupération inférieure à 100 % et a décidé que la validation de l’efficacité de l’extraction pour les essais de détection d’endotoxines n’est pas recommandée (Bryans et al. 2004). Le Sterilization Working Group 8 de l’AAMI (AAMI STWG08, Microbiological Methods) a adopté ces mêmes conclusions au cours des plus récents procédés de révision, et aucune exigence supplémentaire n’a été ajoutée concernant la nécessité de valider l’efficacité de l’extraction (ANSI AAMI ST72).
4.3 Qualification des réactifs
Quelle que soit la technique utilisée (méthodes par gélification, cinétiques ou en point final), la qualification des réactifs reste la même que celles de la Pharmacopée Européenne 2.6.14 et de l’USP <85>.
Les seules différences apportées par la norme concernent les méthodes cinétiques et les méthodes en point final. En effet, il convient que la courbe d’étalonnage ne soit pas supérieure à 4 log, car une courbe de 5 log ou plus peut être interprétée de manière incorrecte dans la partie médiane de la courbe. Si la courbe d’étalonnage est inférieure à 2 log, il est suggéré d’effectuer des dilutions de facteur 2 de la courbe d’étalonnage.
4.4 Qualification de l’analyste : Nouvelle exigence demandée
Il convient de démontrer le maintien des compétences de l’analyste afin de s’assurer qu’un analyste qualifié conserve les compétences et la formation appropriées pour effectuer un dosage, après sa qualification initiale.
Chaque analyste effectuant l’EEB doit démontrer ses compétences en menant à bien la méthode de qualification des réactifs. Ces compétences peuvent être démontrées par :
- La démonstration d’une performance acceptable constante du dosage (tendance)
- L’essai d’aptitude
- La requalification de l’analyste ; et/ou
- L’analyse des tendances des non-conformités.
5. Essais de routine, surveillance et interprétation des données :
Quelle que soit la technique utilisée (méthodes par gélification, cinétique ou en point final), les exigences des essais de routine et du nombre de réplicats restent les mêmes que celles de la Pharmacopée Européenne 2.6.14 et de l’USP <85>.
Cependant, pour les Méthodes par gélification, l’interprétation des résultats est différente de celle de la Pharmacopée Européenne 2.6.14 et de l’USP <85> en cas de résultat positif dans l’un des tubes :
“L’élément soumis à essai est acceptable […] lorsque des résultats négatifs sont obtenus dans les deux tubes contenant la solution d’échantillon.
Si des résultats positifs sont obtenus dans l’un des tubes contenant la solution d’échantillon, lors de l’essai à la DMS, l’élément soumis à essai dépasse la spécification relative aux endotoxines. Se référer aux recommandations relatives à l’investigation associée un résultat de limites hors spécifications (Annexe C)“.
Alors que pour la PE 2.6.14 et l’USP <85>, l’essai devait être répété en cas de résultat positif dans l’un des tubes : “Si l’un des résultats obtenus avec la solution A est positif et l’autre négatif, répétez l’essai. Si, lors de la répétition, les 2 résultats obtenus avec la solution A sont négatifs, la préparation à examiner satisfait à l’essai ; si l’un des résultats obtenus pour la solution A (ou les deux) est positif, la préparation à examiner ne satisfait pas à l’essai.”
6. Maintenance de la méthode EEB / Études d’adéquation, évaluation, réévaluation : Nouvelles exigences de la norme
Afin de détecter les changements involontaires susceptibles d’entraîner des résultats d’essai invalides, une démonstration périodique de l’adéquation continue doit être effectuée si cette adéquation n’est pas systématiquement démontrée par un PPC (Positive Product Control) positif valide utilisant des méthodes cinétiques.
Les contrôles positifs de produit ou PPC de routine de la méthode d’essai cinétique peuvent permettre d’atteindre cet objectif.
Pour les modifications de produits, il convient d’effectuer des évaluations permettant de confirmer que le procédé de fabrication peut produire des produits respectant les limites établies après la modification. Il convient de justifier le nombre d’échantillons/de lots soumis à essai.
Une réévaluation (nouvelle étude d’adéquation) doit être effectuée pour tout changement susceptible de présenter un impact sur l’essai (susceptibles de modifier les niveaux d’endotoxines bactériennes) tels que :
- Modification apportée au produit : introduction de nouveaux matériaux, nouvelle configuration du produit…
- Modification apportée au procédé de fabrication : nouveau process de nettoyage, nouveau traitement de surface, nouveau process de stérilisation (pour les essais de post-stérilisation), site de fabrication différent…
- Modifications apportées à une méthode d’EEB : modifications de la méthode d’extraction, de la technique d’EEB (ex : gélification au lieu de méthode cinétique, utilisation d’un système cinétique colorimétrique à cartouche…), changement de fabricant de lysat, changement de laboratoire, d’analyste, de matériaux ou d’équipements de laboratoire.
Cette (Ré)évaluation doit comporter une évaluation de l’impact de la modification sur le résultat de l’essai. Les résultats de cette réévaluation doivent être consignés, et les études d’adéquation doivent être répétées si nécessaire.
Pour les méthodes cinétiques et les méthodes en point final, si un essai valide est obtenu, y compris un PPC valide, une étude d’adéquation portant sur un lot est considérée comme suffisante.
7. Alternatives aux essais par Lots
Dans la pratique, l’absence d’endotoxines et de pyrogènes en général est confirmée par l’utilisation des essais par lots du produit fini pour la libération de ce dernier. Les alternatives aux essais par lots peuvent être utilisées s’il a été démontré que le procédé de fabrication, les éléments d’entrée du procédé (matériaux, services collectifs) et l’environnement de fabrication sont bien contrôlés et capables de produire des produits dont les niveaux d’endotoxines sont constamment conformes aux limites spécifiées. Elles nécessitent une évaluation des risques qui doit être revue régulièrement (à chaque modification ayant un impact potentiel sur le niveau d’endotoxines) afin d’évaluer les critères utilisés pour établir le plan d’échantillonnage et des données correspondantes démontrant que le procédé de fabrication peut produire un produit respectant systématiquement les limites d’endotoxines spécifiées.
Ces données comprennent généralement l’essai d’un nombre spécifié de lots, l’essai sur une période de temps spécifiée, l’essai de produits principaux représentatifs, l’essai de matières premières/composants et/ou de procédés en cours de fabrication, la vérification des contrôles opérationnels de fabrication, en particulier des eaux de process potentiellement contaminées.
- Les critères pour établir des alternatives aux essais par lots sont :
L’Identification des étapes clés du procédé ou des points de contrôle, ainsi qu’une évaluation supplémentaire des risques visant à démontrer que le procédé de fabrication aboutit à un produit respectant systématiquement les limites d’endotoxines spécifiées.. - La validation, conception du procédé de fabrication, et contrôle (niveaux d’alerte et d’action), ainsi que par un examen périodique et des ajustements effectués si nécessaire.
- La justification documentée et le plan d’échantillonnage défini.
Il convient d’envisager un plan d’échantillonnage en cas de défaillance, ainsi que les exigences nécessaires pour revenir à un plan alternatif aux essais par lots ou à un plan d’échantillonnage réduit, et de les documenter, le cas échéant.
Les alternatives aux essais par lots peuvent comprendre plusieurs options, notamment la réduction du nombre d’échantillons soumis à essai, la réduction de la fréquence des essais, l’essai sur des représentants de familles de produits ou l’utilisation d’alternatives au produit fini, par exemple un produit de substitution.
Le cas où des alternatives aux essais par lots peuvent être envisagées sont les suivants :
- Les produits présentant un historique de contrôle réussi des endotoxines du produit fini,
- Les produits présentant un risque de contamination par des endotoxines plus faible en raison de l’absence d’exposition à une eau qui n’a pas été qualifiée pour contrôler les niveaux d’endotoxines,
- Les produits soumis à un procédé de fabrication qui a été validé pour dépyrogéner le dispositif,
- Les produits qui, sur la base de leur contact prévu avec le patient, présentent un risque attendu mineur ou négligeable de réaction pyrogène.
En cas d’utilisation d’un plan d’échantillonnage alternatif aux essais par lots, il convient d’évaluer l’impact d’un résultat hors spécifications sur des lots non soumis à essai ou sur le produit fini, conformément aux procédures établies pour les produits non conformes. Il convient de tenir compte, dans cette évaluation, du risque pour tous les produits représentés par le plan d’échantillonnage. Dans le cadre de l’investigation, il convient d’examiner les lots libérés auparavant et associés à des alternatives aux essais par lots et d’évaluer les risques qu’ils présentent.
Ces résultats hors spécifications peuvent remettre en question le choix de procéder aux alternatives aux essais par lot. C’est pourquoi, il est important de les consigner dans le plan de gestion des risques qui doit être revu régulièrement afin de prendre les décisions qui s’imposent (revenir aux essais par lots ou adopter un plan d’échantillonnage réduit).
8. Limites d’Endotoxines applicables
Les limites d’endotoxines des DM recommandées par cette norme sont les mêmes que celles fixées par l’USP <161> : Des études menées chez l’homme ont confirmé qu’une dose de 5 UE.kg-1 constituait un seuil de tolérance approprié pour des endotoxines bactériennes en contact avec le système circulatoire ou le système lymphatique. En prenant une masse corporelle de 70 kg, la quantité maximale d’endotoxine pouvant être administrée en une heure est de 350 UE. La limite fixée à l’origine par la FDA et actuellement utilisée par l’USP a été réduite de 350 UE à 200 UE pour tenir compte des inefficacités potentielles de l’extraction, et a fixé la limite de 20 UE/dispositif sur la base du regroupement des extraits de 10 dispositifs (en supposant dans le pire des cas que les 200 UE pourraient provenir d’un seul dispositif dans l’extrait regroupé de 10 dispositifs).
La limite de 2,15 UE/dispositif a été fixée pour les dispositifs en contact avec le liquide céphalo-rachidien, car l’endotoxine dans l’espace intrathécal (ou intrarachidien) a un pouvoir pyrogène beaucoup plus important que l’endotoxine avec contact intravasculaire.
Pour les dispositifs qui entrent directement ou indirectement en contact avec l’environnement intraoculaire, une limite d’endotoxines inférieure peut s’appliquer.
Des limites d’endotoxines plus élevées que 20 UE/dispositif peuvent être justifiées et nécessitent une acceptation par les autorités réglementaires.
Par exemple, une limite de 35 UE/dispositif ou plus pourrait être appropriée pour les dispositifs médicaux sans exposition systémique, étant donné que la puissance de l’endotoxine pour stimuler une réaction pyrogène est plus faible et qu’il n’est pas nécessaire d’ajouter une réduction supplémentaire de la limite en raison des inefficacités potentielles d’extraction.
Les produits de santé implantables sous-cutanés sans exposition systémique sont des exemples de produits pour lesquels des limites d’endotoxines plus élevées peuvent être justifiées.
9. Recommandations relatives aux résultats de limites hors spécification (OSL) et aux investigations associées
Les OSL, Out of Spécifications Limits ou limites hors spécification représentent les échantillons dont le résultat de l’EEB est valide, mais qui dépassent la spécification de limite d’endotoxines du produit.
Les recommandations de l’annexe C de la nouvelle norme spécifient que :
- Si l’essai d’investigation complémentaire révèle une erreur du laboratoire, sous forme d’une contamination qui aurait pu se produire pendant l’extraction ou pendant l’EEB initial, l’essai initial peut être considéré comme non valide (c’est-à-dire un “no-test”) et l’EEB peut être répété en utilisant de nouveaux échantillons de produit et la taille d’échantillon initiale,
- Si l’investigation du laboratoire ne permet pas d’identifier la cause profonde de l’OSL, des essais d’investigation supplémentaires (par exemple, un contre-essai de l’extrait) visant à vérifier la validité du résultat initial peuvent être effectués afin d’examiner la possibilité qu’une contamination extrinsèque se soit produite pendant l’essai initial d’endotoxines bactériennes (=contamination provenant de sources autres que les matières premières du produit, les auxiliaires technologiques, le traitement du produit ou l’emballage du produit).
Par ailleurs, un contre-essai de l’extrait doit être réalisé en utilisant les préparations d’extraction originales afin d’examiner la possibilité d’une contamination extrinsèque lors de l’EEB initial.
Il convient de soumettre à essai au moins deux fois (2x) le nombre de réplicats de la préparation d’extraction de l’échantillon original utilisée lors de l’essai initial. Par exemple, dans un essai initialement réalisé en un seul essai en double exemplaire, le contre-essai d’investigation consisterait en deux essais, chacun réalisé en double exemplaire.
Si l’extrait confirme la présence d’endotoxines, un essai répété du produit utilisant de nouveaux échantillons de produit peut être effectué afin d’examiner la possibilité d’une contamination extrinsèque survenue lors de la préparation initiale de l’extraction de l’échantillon. Dans un essai répété de produit, il est recommandé d’augmenter le nombre d’échantillons de produit par rapport au nombre initial soumis à essai. Par exemple, il convient de soumettre à essai au moins deux fois (2x) le nombre d’éléments soumis à essai initialement lors de l’essai répété du produit.
Conclusion
L’impact majeur apporté par cette nouvelle norme est l’obligation de réaliser les EEB sur chaque lot de produit finis (Essais par Lots) à moins de recourir aux alternatives aux essais par lots sous réserve de réunir des conditions bien définies, justifiées et documentées.
Il est possible d’utiliser les courbes d’étalonnage archivées lors de la validation des méthodes d’essais pour les méthodes cinétiques et en point final. Il est à souligner que la validation de l’efficacité de l’extraction des endotoxines n’est pas demandée dans cette norme et même non recommandée.
On peut cependant regretter que la version actuelle ne reflète pas l’évolution des réglementations nationales qui régissent les EEB et que des méthodologies alternatives comme le facteur C recombinant (rFc), aient été exclues. La méthode rFc représente en effet la dernière solution de pointe pour tester efficacement les endotoxines bactériennes en réunissant de nombreux avantages : méthode plus spécifique en évitant les faux positifs dus aux β-glucanes, méthode validée selon les critères standards des tests d’endotoxines bactériennes de la PE (Chap. 2.6.32), utilisation de recombinants qui garantissent la cohérence de lot à lot, méthode qui s’affranchit de l’utilisation des Limules et contribue ainsi à la préservation de ces espèces menacées, en concordance avec la Directive 2010/63/UE relative à la protection des animaux utilisés à des fins scientifiques (règle des 3R). L’enquête publique en France, dont les résultats ont été publiés le 25/03/2022, a d’ailleurs recueilli une majorité d’oppositions au projet actuel en raison de l’exclusion de la méthodologie du rFC.
Cette méthodologie rFC ne sera, à l’heure ou cet article est écrit, pas intégrée dans le nouveau projet de norme, en raison du refus des Etats Unis.
Partager l’article

Références
- WEBER, J., RYAN, G., BERLAM, S. Implementation of a rapid endotoxin testing platform. American Pharmaceutical Review, 17 (Endotoxin Detection Part II Supplement): p. 12-15. (2014)
- BRYANS T.D., BRAITHWAITE C., BROAD J., COOPER J.F. et al. Bacterial Endotoxin Testing: A report on the methods, background, data, and regulatory history of extraction recovery efficiency. Biomed. Instrum. Technol. 2004, 37 pp. 73–78
- ANSI/AAMI ST72 :2019 : Bacterial endotoxins— Test methods, routine monitoring, and alternatives to batch testing
- PE 2.6.14 : Essais des endotoxines bactériennes
- USP <85> : BACTERIAL ENDOTOXINS TEST
- USP <161> MEDICAL DEVICES: BACTERIAL ENDOTOXIN AND PYROGEN TESTS – ISO 11737-1: 2021 : Stérilisation des produits de santé – Méthodes microbiologiques – Partie 1 : détermination d’une population de micro- organismes sur des produits
Acronymes & Définitions
AAMI: American Association of Medical Instrumentation
DM : Dispositif Médical
DMS : Dilution Maximale Significative ou maximum valid dilution (MVD), dilution maximale pour un échantillon ou volume total d’extraction utilisé par rapport à la sensibilité d’un EEB dans lequel la limite d’endotoxines spécifiée peut être détectée EEB: Essais des Endotoxines Bactériennes
FDA: Food & Drug Administration
LAL : Lysat d’Amébocytes de Limule, réactif extrait d’amébocytes prélevés dans l’hémolymphe de la limule, Limulus polyphemus, qui réagit avec l’endotoxine pour former un caillot gélatineux et qui est utilisé pour estimer les niveaux d’endotoxines dans les méthodes d’EEB
MAT : Monocyte Activation Test (Essais d’Activation des Monocytes)
OSL : Out of Specifications Limits (limites hors spécifications), échantillon dont le résultat de l’EEB est valide, mais qui dépasse la spécification de limite d’endotoxines du produit
PE: Pharmacopée Européenne
PPC : Product Positive Control (contrôle positif du produit), échantillon avec surcharge d’une quantité connue d’endotoxine utilisé pour confirmer que le produit soumis à essai ne présente pas de facteurs d’interférence
rFC: recombinant Factor C (Facteur C recombinant)
UE : unité d’endotoxine
USP : United States Pharmacopeia (Pharmacopée Américaine)
Céline PEREZ, Pr Edith FILAIRE, Dr Christiant POINSOT – Groupe ICARE
Voir le profil sur ![]()
