


Sommaire
- L’intérêt de la désinfection automatisée des surfaces d’un RABS par decontamination aérienne
- Utilisation d’isolateurs individuels pour la thérapie cellulaire autologue
- Calculation of greenhouse gas (GHG) emissions expressed in CO2eq of an open system (AinB) compared to a closed system equipped with isolators (AinD)
- Exigences croisées de la norme ISO 13408-2 et de l’Annexe 1
- Employing Conductivity Measurements for On-site Residue Quantification
- LEAN. Utiliser le digital pour transmettre et former vos équipes sur le terrain
- Deciphering the complex characteristics of nanomedicines
- L’industrie pharma doit réduire sa trace carbone, ... le traitement d’air. Part 2
Exigences croisées de la norme ISO 13408-2 et de l’Annexe 1
La filtration stérilisante est une étape cruciale dans les procédés aseptiques. Elle permet de garantir la stérilité des produits, donc d’assurer la sécurité des patients. Dans un contexte réglementaire en constante évolution, il est impératif de se conformer aux nouvelles exigences. Cet article présentera la démarche générale de qualification des filtres et de validation du procédé de filtration stérilisante, en synthétisant les exigences normatives (ISO 13408-2) et réglementaires (Annexe 1 des BPF).

En éliminant les micro-organismes des produits liquides et des gaz, la filtration stérilisante joue un rôle essentiel dans la production pharmaceutique. Elle est utilisée sur des produits qui sont sensibles aux procédés de stérilisation conventionnels (chaleur sèche ou humide, rayonnement ionisant ou stérilisation ETO). Or, l’efficacité de l’élimination microbienne par filtration dépend de nombreuses variables, qui si elles sont mal maîtrisées, rendent inefficace ce procédé.
Un critère essentiel dans le choix d’un filtre est donc sa capacité à retenir les particules indésirables (microorganismes et particules). La taille nominale des pores doit être suffisamment petite pour retenir les contaminants, tout en permettant un débit adéquat du produit. Pour cela, il est important de choisir un filtre avec une taille de pores appropriée, généralement de 0,22 micron, compatible avec le produit à filtrer (ne devant pas interagir avec le produit ni altérer ses propriétés physiques ou chimiques). Il est donc important de choisir un filtre fabriqué à partir de matériaux inertes. Le filtre, ainsi que le système de filtration doivent résister aux méthodes de nettoyage et de stérilisation utilisées dans le procédé de fabrication.
Pour terminer, les filtres doivent maintenir leur intégrité structurelle et leurs propriétés de filtration sous des conditions opérationnelles (pression différentielle, débit, durée) et thermiques extrêmes, sans compromettre la stérilité du produit. La sélection du système avec une résistance mécanique et chimique élevée, ainsi qu’une durée de vie suffisante doit garantir une filtration efficace durant la production. Le système doit aussi permettre le monitoring en continu des paramètres critiques et des tests d’intégrité pré- et post-filtration in situ. En somme, le choix du filtre stérilisant doit être basé sur des critères rigoureux pour assurer l’efficacité de la filtration stérilisante et la qualité du produit final.
“La filtration stérile est donc une étape cruciale de la production, et la validation de ce processus est donc essentielle.”
Les questions les plus fréquemment posées dans l’industrie sont les suivantes : Quels sont les critères de sélection du filtre ? Quels sont les paramètres critiques du processus de filtration ? Qualifier un filtre et valider la filtration ! Quelles sont les différences ? Comment démontrer l’efficacité de l’élimination microbienne par l’épreuve bactérienne ? Avec les tests d’intégrité en PUPSIT, quels sont les autres éléments de contrôle qui doivent être mis en place en routine ?
La validation du filtre vise à prouver que le processus élimine systématiquement les microorganismes d’un fluide, sans nuire à la qualité du filtrat. La conformité aux exigences normatives et réglementaires, de qualification / validation, doit garantir que le processus de filtration est fiable et reproductible. Contrairement à d’autres procédés de stérilisation, où les paramètres sont surveillés en continu, la rétention microbienne et l’intégrité physique du filtre ne peuvent pas être monitorées en continu, sur toute la durée de la filtration. La surveillance continue ne se réalise que sur des paramètres qui sont corrélés indirectement à la rétention microbienne, tels que le débit, la pression, le volume. D’où l’importance de valider selon les principes normatifs, cet aspect fiabilité et reproductibilité, afin de prouver l’interdépendance des paramètres de surveillance avec l’intégrité physique du filtre et sa capacité de rétention microbienne.
Heureusement, la norme ISO 13408-2, ainsi que les paragraphes 8.79 à 8.95 de l’Annexe 1 définissent clairement les étapes de qualification, validation et surveillance qui doivent s’appliquer. Dans votre stratégie de validation, il faudra également prendre en considération les orientations de l’Annexe 1 applicables aux Single Use System (SUS) sur les éléments critiques du système de filtration, dont le filtre. Comprendre les variables critiques de la filtration stérilisante, et les préconisations réglementaires est essentiel pour s’assurer que le procédé est correctement défini et validé. En outre, les défis sont nombreux : les conditions en durée, températures ou pression qui règnent sur la chaîne de production peuvent être extrêmes, les composés mis en œuvre sont parfois corrosifs, visqueux, fragiles ou réactifs. Autant de contraintes à intégrer dans votre stratégie de validation pour obtenir des produits sûrs, efficaces, stériles, dans des délais raisonnables.
Pour les procédés de stérilisation classiques (chaleur sèche ou humide, rayonnement ionisant…), la cinétique d’inactivation microbienne suit une loi mathématique permettant de calculer un niveau de stérilité garanti. L’élimination des organismes d’un fluide par filtration ne suit pas cette décroissance mathématique. L’ensemble des exigences réglementaires soulignent donc l’importance de comprendre la nature de la charge biologique initiale d’un fluide. Outre de connaître la concentration et le type de germes présents dans le produit, il est essentiel de déterminer la taille et la forme de ces microorganismes. Il faut comprendre l’influence du fluide à filtrer, et du procédé de fabrication avant filtration, sur la biocharge (type, concentration, taille, forme), notamment son impact sur le micro-organisme d’essai utilisé pour valider cette capacité de rétention microbienne.
Dans ce processus complexe de validation, où chaque étape doit être minutieusement respectée, de nombreuses expertises interviennent à différents niveaux. La constitution de votre équipe de travail sera d’une importance cruciale ! Il faudra vous entourer à minima d’un expert microbiologiste (pour la compréhension de l’impact produit sur la biocharge), d’un expert produit (pour la compréhension de l’impact filtration sur votre produit), d’un expert filtration (pour la conception et qualification de votre système de filtration) et d’un expert en assurance de stérilité (pour la validation du procédé aseptique).
1. Caractérisation du filtre et du système
La première étape est la caractérisation du filtre et du système de filtration (montage, connexion…). La caractérisation des filtres stérilisants consiste à sélectionner les filtres aptes à être utilisés, ainsi qu’à justifier les paramètres procédés et les limites de tolérances. Il faut donc au préalable, réaliser une analyse de risque justifiant vos choix techniques et qualités.
Un rationnel expliquera le choix du filtre, le fournisseur de filtre, l’équipement de filtration, ainsi que l’ensemble des paramètres critiques et déterminera :
- Les variables impactant l’efficacité de l’élimination microbienne (Table 1)
- Les limites de tolérances de ces variables
- La conception du système de filtration (filtre unique, en parallèle ou en série – double ou redondant)
- L’emplacement des filtres dans le procédé (le dernier filtre stérilisant étant le plus proche possible du point de remplissage aseptique)
- Les risques de perte de stérilité lors de la réalisation des tests d’intégrité en PUPSIT (Après Stérilisation Avant Utilisation). En effet, le système doit permettre la réalisation des tests d’intégrité, avant et après filtration en place, sans engendrer un risque de contamination microbienne.
- Les risques associés à l’externalisation (SUS, achat de filtre stérile…).
- Les effets du produit sur le filtre et du filtre sur le produit.
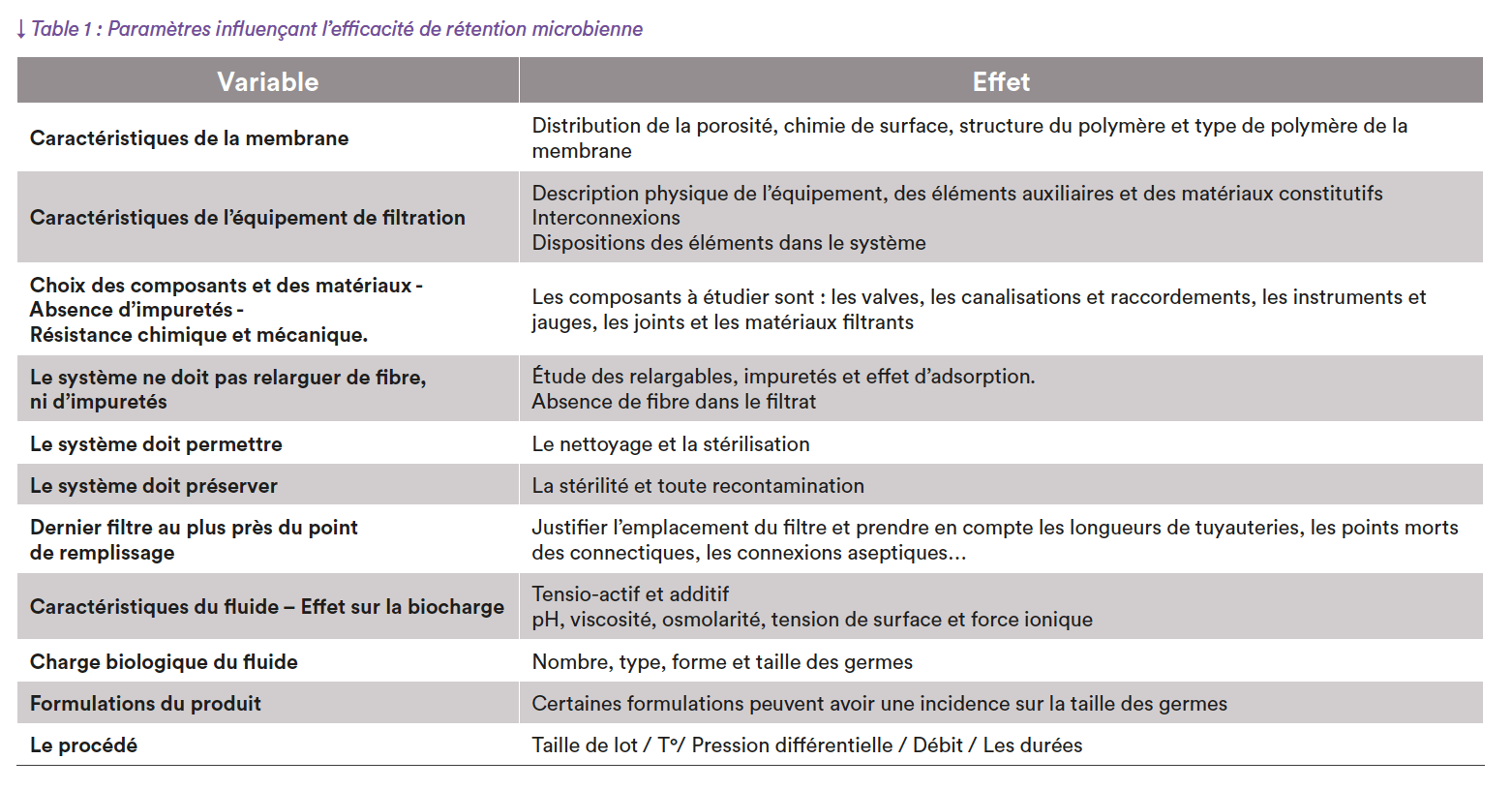
Une étape dans cette analyse de risque est donc la caractérisation du procédé de filtration, de ses paramètres critiques et des moyens de surveillance. L’établissement des paramètres critiques et des tolérances étant basé sur 3 principes :
- Le choix du filtre et de ses caractéristiques techniques.
- La connaissance du produit à filtrer et de sa biocharge, afin d’obtenir un filtrat stérile sans compromettre la sécurité, la qualité, l’efficacité et la performance du produit.
- La sélection d’une méthode de nettoyage et de stérilisation pour le système de filtration.
2. Sélection du filtre
Le choix d’un filtre va dépendre de plusieurs facteurs. Sa capacité de rétention microbienne qu’il faudra valider pour chaque combinaison filtre stérilisant /produit, mais aussi, les effets du filtre sur l’éluat. Pour cela il faudra étudier les relargables, impuretés que le filtre libère dans le produit. Mais également, évaluer les effets de l’adsorption des composants du produit sur le filtre, surtout sur la qualité du filtrat. Pour rappel, l’adsorption est un mécanisme où les composants du produit se lient au matériau filtrant modifiant la composition du filtrat. Les facteurs influençant cette adsorption, qui seront à justifier (certains à valider), sont repris en figure 1. La plupart seront ensuite monitorés en routine. Pour cela, des limites de tolérances hautes et basses seront à spécifier lors de la validation.

3. Caractérisation du filtre
Le choix du type de filtre (marque et fournisseur), sa taille et ses constituants sont primordiaux. Il doit être adapté au fluide à stériliser, ainsi qu’au procédé de fabrication et au système de filtration utilisé.
C’est à l’utilisateur de démontrer la pertinence de ses choix, via un rationnel qui abordera :
- la surface utile du filtre
- la porosité du filtre – la compatibilité thermique du filtre avec les conditions opératoires, dont les températures de stérilisation
- la force hydraulique nécessaire pour résister à la pression différentielle du procédé
- la configuration du filtre (plaque ou cartouche ; filtre redondant…)
- la durée de vie du filtre
- la charge biologique (type, taille et concentration des germes à éliminer).
4. Caractérisation de l’équipement de filtration
Concomitamment à la sélection de votre filtre, il faudra caractériser votre système de filtration et fixer les performances de votre procédé. Dans un 1er temps, il faudra faire des choix techniques. À cette étape, il est judicieux de vous faire aider de l’expertise des fournisseurs de filtre qui vous conseilleront sur l’adéquation des composants de votre équipement (filtre, carter et corps de filtre) avec votre procédé. Il vous faudra réaliser une description physique précise de votre équipement et des éléments auxiliaires, dont les matériaux constitutifs du système. Il faudra justifier le choix de chaque composant, leur interconnexion et leur disposition. Bien entendu, aucun de ses éléments ne devra contaminer le produit. C’est-à-dire que les études des relargables, extractibles et adsorption concernent le système et non pas uniquement le filtre. Il faudra démontrer que les composants du système ne transfèrent pas d’impuretés ni ne modifient la qualité du produit. En plus du filtre, les composants critiques d’un système de filtration sont :
- les systèmes de canalisation et de raccordement
- les valves
- les jauges et autres instruments de mesure
- les joints et les garnitures
- les matériaux filtrants
- les cages de protection ou carter
- les instruments de surveillance
En plus d’apporter l’assurance de la stérilité du filtrat, l’étude de conception garantira la maîtrise et la mesure des paramètres procédé (débit, pression, température, durée). Elle justifiera le choix des capteurs, ainsi que l’emplacement de ces dispositifs de monitoring. Elle intégrera une évaluation des risques justifiant l’emplacement du filtre minimisant le risque de recontamination après filtration. Pour cela, des systèmes simples sont à privilégier, avec un minimum de joints et de garnitures, ainsi que de placer le filtre stérilisant au plus près du dispositif de remplissage. Le système de filtration devant être stérile, votre étude de conception tiendra compte des contraintes de nettoyage et de stérilisation (stérilisation en place ou stérilisation chez le fournisseur puis assemblage aseptique). La maîtrise de vos fournisseurs, ainsi que la compréhension des risques engendrés par les SUS sera une nécessité !
Enfin, la conception évaluera le risque de réalisation des tests d’intégrité avant / après utilisation, en place (PUPSIT).
Ces décisions qualité seront décrites dans un rationnel type analyse de risque. Comprendre les systèmes de filtration et leurs composants peut se révéler compliqué, étant donné la variété des options disponibles. La conception et la qualification, selon les principes QbD (Quality by Design), des éléments constitutifs du système offriront un haut niveau d’assurance de stérilité.
5. Le fluide (le produit) à filtrer
Certaines caractéristiques du produit ont une influence sur l’efficacité de la filtration, d’autres sont à conserver pour garantir sa qualité. Ses paramètres sont à prendre en compte dans votre stratégie de validation, et vérifier, pour chaque attribut, leur maintien dans des limites prédéfinies (Table 2).

La norme ISO 13408-2 insiste sur l’effet potentiel de la formulation du produit et de la nature du filtre sur la taille cellulaire des microorganismes. Un expert microbiologiste étudiant ces paramètres sera un atout majeur lors de la construction de votre analyse de risque et sur l’analyse des résultats de validation. Il faudra statuer sur le fait que votre système de filtration est apte à arrêter la biocharge que votre produit et process favorisent.
Pour connaître la charge biologique de votre produit, la réglementation impose de faire un prélèvement pour analyse de la biocharge sur chaque lot, avant la filtration stérilisante. En validation, il faudra, en plus, identifier les espèces microbiennes et analyser l’influence de votre produit sur leurs tailles. En lien avec cette biocharge, il est essentiel de définir les conditions de stockage (température, système de protection de la contamination tel que inertage qui consiste à remplacer l’atmosphère d’un espace donné, tel qu’un réservoir de stockage, une cuve ou un espace clos, par un gaz inerte afin d’empêcher la contamination, l’oxydation ou d’autres réactions indésirables), ainsi que de fixer un délai entre la dernière étape de fabrication et le début de la filtration. Cette durée influence le développement microbien. La durée totale de filtration est également un facteur à définir puis à valider. Les paramètres températures et durée, étant des facteurs de proliférations microbiennes, ils doivent être clairement validés puis respectés en routine. Bien entendu, les méthodes de détermination de la charge biologique respecteront la norme ISO 11737-1 ou la monographie PE 2.6.12.
Lors de vos lots de routine, votre système qualité devra prouver la maîtrise de cette charge biologique, dans le produit, avant filtration. Pour cela, les préconisations de l’Annexe 1 des BPF seront à implémenter et à respecter.
6. Comptabilité filtre / fluide
En parallèle de l’étude des effets de votre produit sur la biocharge, il faudra initier des études de compatibilité Filtre /Fluide, afin de connaître les effets que le filtre peut engendrer sur votre produit et aussi de vérifier l’absence d’impact de votre produit sur sa capacité de rétention. C’est à l’utilisateur de démontrer cette compatibilité via les essais suivants :
- Impacts de la formulation du produit, ainsi que les effets du procédé (température, durée, débit), sur les attributs chimiques, physiques et la performance du filtre
- Effets du filtre sur les attributs biologiques, chimiques et physiques du fluide. Cette analyse intégrera les études des extractibles, des relargables, les endotoxines, la création de particules ou la libération de fibres, les études d’adsorption. L’identité et la quantité des relargables et impuretés devront être déterminées sur le produit à filtrer. Si votre étude utilise un produit de substitution, une justification sera à apporter. Enfin, les études devront démontrer l’absence de toxicité des extractibles provenant du filtre. La mise à disposition, par les fabricants de filtres, de données sur les extractibles ou sur les compatibilités chimiques, permettra d’alimenter votre analyse.
Pour résumer, les exigences réglementaires imposent dans un 1er temps de caractériser le filtre et le système de filtration. Lors de ces étapes de qualification, une évaluation des risques justifiant vos choix techniques et qualité sera élaborée. Définir précisément, également, vos paramètres procédés, fixer des limites et élaborer votre stratégie de surveillance (par le choix et l’emplacement de vos capteurs), feront partie intégrante de votre stratégie de qualification.
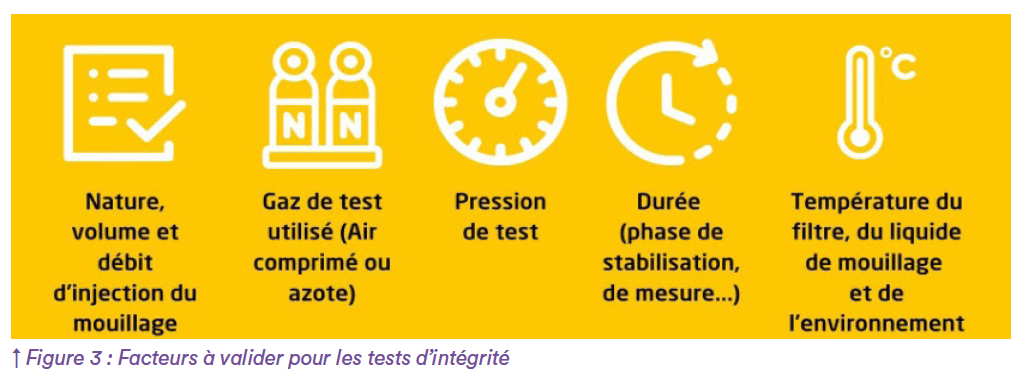
Pensez, en outre, qu’il faudra également définir vos paramètres concernant vos tests d’intégrité (Figure 3). Après cette phase de conception, des études seront à mener pour vérifier les effets (ou l’absence d’effet) du filtre sur le produit et du produit sur le filtre.
Lorsque ce 1er travail aura été achevé, vous démarrez ensuite vos essais de validation, dont le but sera de démontrer la fiabilité, l’efficacité et la reproductibilité du procédé de filtration. Pour cela, selon que le fluide à filtrer soit un gaz ou un liquide, 3 ou 5 étapes sont recommandées (figure 2).
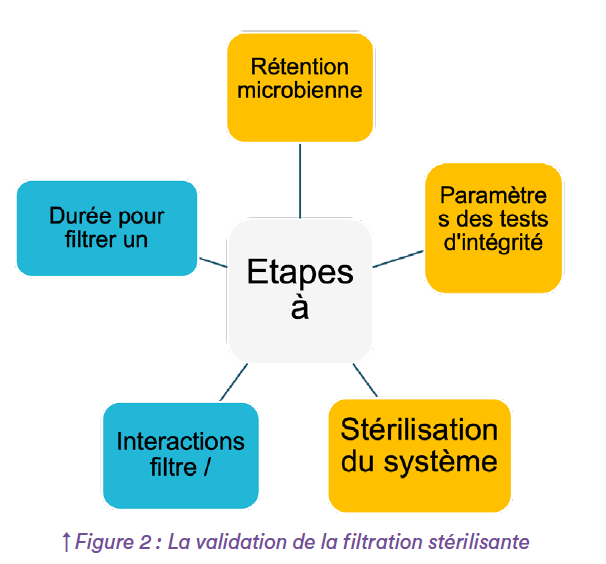
7. Epreuve bactérienne
Cette étape importante dans la stratégie de validation permet de garantir que la membrane et la cartouche de filtration possèdent les critères de performance d’un filtre stérilisant, c’est-à-dire que la membrane doit retenir physiquement TOUS les microorganismes (bactéries, champignons et protozoaires) d’un fluide en produisant en aval, un effluent stérile. Cela consiste à évaluer l’aptitude du filtre à retenir les organismes de références d’une suspension bactérienne liquide en simulant les conditions de process worst-case. Il est à noter que les virus ne sont pas totalement filtrés et ne sont pas concernés par les exigences de la norme ISO 13408-2.
La filtration stérilisante des gaz, avec des filtres hydrophobes, est généralement validée par le fournisseur du filtre en utilisant un aérosol d’essai. Selon la littérature scientifique et normative, l’influence du gaz porteur est peu significative sur le procédé de filtration, ce qui permet d’éviter d’initier un test d’épreuve bactérienne spécifique à votre procédé. Votre obligation sera juste d’évaluer les données de rétention générées par le fabricant afin de garantir leur applicabilité par rapport à vos conditions opérationnelles.
Concernant l’épreuve bactérienne, sur filtre hydrophile pour la stérilisation des liquides, le test de rétention doit obligatoirement simuler les conditions de production les plus contraignantes. Idéalement il faudrait réaliser une épreuve bactérienne pour chaque trio Filtre / Produit / Conditions opérationnelles, c’est-à-dire que le fluide d’essai doit être le produit à filtrer pour chaque condition opératoire. Dans la réalité et pour des raisons de délai et de coût, il est possible de regrouper les liquides ayant des propriétés similaires. Bien entendu, les représentants worstcase seront utilisés pour l’étude, avec un rationnel écrit supportant la justification du trio représentatif utilisé.
Si le fluide à filtrer ne peut pas être utilisé pour des raisons de propriétés anti-microbiennes ou de conditions opérationnelles inhibitrices de croissance microbienne (Température, durée, pH…), vous avez la possibilité d’utiliser un fluide ou des conditions de simulation, se rapprochant le plus de votre réalité opérationnelle, en respectant les attributs de la Table 2. Chaque modification sera à justifier. Vous pouvez bien entendu éliminer le composé anti-microbien de votre produit, mais aussi modifier les éléments suivants : pH, température du fluide durant l’épreuve, durée réduite d’exposition du microorganisme de référence au fluide, utilisation d’un germe résistant aux propriétés du fluide.
“Lors de vos lots de routine, votre système qualité devra prouver la maîtrise de cette charge biologique, dans le produit, avant filtration.”
Deux micro-organismes de référence sont définis dans la norme ISO 13408-2 pour être utilisés dans le procédé de validation. Il s’agit de Brevundimonas diminuta (ATCC 19146, pour les filtres de 0,22 µm) et d’Acholeplasma laidlawii (ATCC 23206, pour les filtres de 0,1 µm retenant les mycoplasmes). Ce dernier doit également être utilisé lorsque la biocharge de votre produit contient des micro-organismes plus petits que Brevundimonas diminuta.
Le niveau d’épreuve minimale est de 10⁷ germes viables/ cm² de surface utile du filtre, dans les conditions de process, avec le produit à filtrer, sur la durée maximale. Le résultat doit être l’obtention d’un filtrat stérile, sur 3 essais consécutifs. Cela permettra de définir la durée maximale de filtration autorisée et le temps total durant lequel le filtre doit être en contact produit.
La difficulté dans ce genre d’essai est de maîtriser l’ensemble des paramètres ayant une influence sur la taille cellulaire et d’avoir un nombre suffisant de données pour connaître le type de germe que sélectionne votre produit, pour cela, l’aide d’un expert microbiologiste vous sera précieuse. Les conditions de culture seront également à définir afin d’obtenir des germes de petites tailles. Outre les facteurs indiqués en table 1, il faudra être attentif à la présence de substances impactant le passage des germes à travers la membrane telle que les liposomes ou la présence d’organismes pléomorphes (mycoplasmes, leptospires ou forme L des bactéries dans une solution de pénicilline).
Grâce aux données de l’épreuve bactérienne, vous établirez des spécifications pour vos tests d’intégrités non destructifs. En effet, la validation est censée établir un lien de reproductibilité entre la rétention bactérienne et l’intégrité physique des filtres. En routine, la réussite de vos tests d’intégrité, en place, avant et après utilisation, vous permettra de prouver la réussite de la stérilisation.
8. Test d’intégrité
Valider et réaliser en routine des tests d’intégrité, c’est fixer les paramètres influençant les résultats de ces tests de performances fonctionnelles (voir Figure 3). Pour rappel, il existe 3 types de tests non destructifs : le point bulle, la diffusion et le test d’intrusion à l’eau. Les deux premiers, conviennent aux filtres hydrophiles et hydrophobes. Le dernier test convient uniquement aux membranes hydrophobes. Après avoir expliqué le choix d’un des 3 types de tests, vous devrez valider les spécifications d’échec ou de réussite du test, qui dépend des paramètres repris en Table 3.
 Un rationnel écrit expliquera chacun de vos choix techniques. En routine, il faudra apporter la preuve que ces facteurs ont été respectés, sinon, le résultat de votre test ne correspondra pas à la réelle intégrité de votre membrane.
Un rationnel écrit expliquera chacun de vos choix techniques. En routine, il faudra apporter la preuve que ces facteurs ont été respectés, sinon, le résultat de votre test ne correspondra pas à la réelle intégrité de votre membrane.
La réalisation des tests d’intégrité avant et après filtration est une obligation pour les filtres liquides (après utilisation pour les filtres à gaz). Les directives réglementaires insistent sur l’importance de tester le filtre en place, dans son boîtier, afin de vérifier l’étanchéité et l’intégrité structurelle du système avant utilisation et l’intégrité de la membrane durant le procédé de filtration.
9. En routine
Les variables influençant la capacité de rétention étant nombreuses et interdépendantes, le monitoring des paramètres critiques est une obligation (voir Table 3). Cette surveillance sera une preuve documentée démontrant que le procédé est maintenu dans les limites de tolérances. Il vous faudra de plus, mettre en place un système maîtrisant et mesurant la biocharge de votre produit avant la filtration.
Enfin, la validation de la filtration stérilisante est un prérequis à la validation de votre process aseptique, dont l’APS (test de simulation aseptique, Aseptic Process Simulation) est une des composantes. La maîtrise de ce système complexe vous apportera une plus grande confiance dans la robustesse et l’assurance de stérilité de votre procédé.
Conclusion
La filtration stérilisante est une étape critique, dans un procédé aseptique. Elle est soumise à des exigences de plus en plus strictes qui soulignent l’importance de la validation de ce procédé. La multiplicité des variables, des attributs et des paramètres à contrôler est importante. La compréhension des facteurs critiques et le respect strict de chacune des étapes de validation vous apporteront la meilleure assurance de stérilité. De la sélection de votre filtre jusqu’à la validation par l’épreuve bactérienne, en passant par l’analyse des interactions filtres / produit, les expertises à développer sont nombreuses. Il est donc important de mettre en place des protocoles rigoureux et de vous appuyer sur la norme ISO 13408-2 qui borde bien le sujet.
Références
1. ISO 13408-2
2. Annexe 1 § 8.79 à 8.95 (Filtration stérilisante)
3. Annexe 1 § 8.131 à 8.139 (Single Use System)
4. Annexe 1 § 8.127 à 8.130 (Closed system)
5. PDA 26 (guide technique de la FDA sur la filtration stérilisante)
6. PDA 36 (guide technique FDA sur la validation aseptique)
7. PDA 44 (guide technique FDA Quality Risk management for aseptic process
Partager l’article


