


Sommaire
- Zero CFU : un objectif et une illusion. Une approche holistique de la maitrise de la contamination
- Part 2: Turning constraints into opportunities to accelerate Sterility Assurance performance
- Key Elements of a Successful Cleaning and Disinfection Program
- Améliorer la maîtrise de la contamination grâce à l’analyse de risque: un pilier de la stratégie CCS selon l’ICH Q9 et Q10
- The control of surfaces in cleanrooms: Questions & Answers
- Methods to Validate Disinfectants
- Clés du succès d’un projet de mise en place d’une solution de nettoyage GMP
- Nouvelle venue dans le monde de la désinfection ?
- Arrêté sècheresse : impacts & opportunités pour l'industrie pharma
Zero CFU : un objectif et une illusion. Une approche holistique de la maitrise de la contamination.
Quand une contamination est trouvée en grade A, une investigation démarre et c’est souvent un exercice long et difficile. Parfois une cause probable est identifiée mais très rarement une cause racine certaine. Néanmoins, un CAPA est mis en place et les opérations de production peuvent reprendre. Et puis quelques temps après, une nouvelle excursion est découverte.
Cela veut-il dire que le CAPA était inefficient ou que la cause de la contamination précédente n’était pas la bonne ?

Quelle que soit la réponse, le site de production ou l’organisation qualité peuvent être mises en difficulté pour leur capacité à conduire une investigation de manière efficace (“Root Cause Analysis deficiencies”) ou que les CAPAs identifiés ne sont pas efficients (CAPA effectiveness check).
Cet article va tenter d’apporter un éclairage nouveau pour aller vers une maitrise pérenne de contamination.
1. Les limites du controle de l’environnement
Historiquement, on considérait que la surveillance microbiologique comme les monitorings environnementaux (EM) permettait de montrer, voire de démontrer la bonne maitrise des opérations aseptiques.
Or, le monitoring environnemental n’offre que des informations très limitées.
D’abord la capacité des méthodes classiques (milieux de croissance en gélose ou liquide pour les APS) à faire pousser un microorganisme potentiellement vivant en grade A est très faible. Les taux de recouvrement sont difficiles à déterminer avec précision car ils dépendent de nombreux facteurs (nature du microorganisme, état de viabilité, etc.), mais on sait qu’ils sont en général faibles, voire très faibles.
Par ailleurs, les prélèvements sont spatialement limités. Si une gélose contact est négative à une localisation donnée cela ne veut pas dire que toute la surface que le prélèvement est censé représenter est dépourvu de microorganisme. Il en est de même pour les prélèvements d’air.
D’autre part, les contrôles de l’environnement sont ponctuels dans le temps. En grade A, les comptages particulaires sont continus mais il n’en est pas de même pour les contrôles microbiologiques avec une capacité de “capturer” tous les événements qui est statistiquement très limitée.
Compte tenu du faible niveau d’informativité du contrôle de l’environnement, si une contamination est trouvée, combien d’autres micro-organismes sont présents ?
A l’inverse s’il n’y a pas de micro-organisme détecté, comment peut-on affirmer l’asepsie de la zone ? Ce sont là les limites des contrôles de l’environnement qui peuvent produire l’illusion que la zone est dépourvue de contaminants. L’absence de détection de microorganisme en Grade A est bien une attente mais également une illusion.
La métaphore du pêcheur est bien connue. Ce n’est pas parce qu’un jeune pécheur n’attrape pas de poisson qu’il n’y pas de poissons dans l’étang. Mais plus il y a de poissons dans l’étang, plus la probabilité que le jeune pécheur en attrape est élevée.

Trop souvent l’industrie s’appuie sur ces contrôles environnementaux pour assoir sa stratégie de construction de la maitrise de la contamination. Cette posture n’est plus admise, en particulier depuis la publication de la dernière révision de l’Annexe 1 de l’EudraLex en 2022.
La maitrise de la contamination repose en réalité sur une approche holistique qui intègre les différents éléments permettant de construire une politique de maitrise basée sur le design des systèmes de barrière, les processus de réduction de la bio-charge et sur leur mise en œuvre.
2. La protection du “sanctuaire”
Les principes de la production aseptique sont conceptuellement très simples mais en même temps très complexes dans leur mise en œuvre.
Il s’agit d’atteindre un niveau de contamination microbiologique nul (le “0 CFU”) et particulaire très faible (< 100 particules de 0,5 microns par pied cube) dans la zone ou le produit et les contenants sont exposés : le Grade A, que nous appelons le “sanctuaire aseptique”.
Comme dans l’antiquité égyptienne où le sanctuaire de la tombe du pharaon était une salle interdite protégée par une série de salles adjacentes à l’accès de plus en plus restreint, le Grade A est une zone ou l’intervention humaine n’est pas souhaitable et que l’on protège par une série de salles propres de propreté croissante.
Le défi est de taille : en effet, la bio-charge à l’extérieur de l’usine de fabrication est très élevée (des millions de particules et de micro-organismes par mètre carré ou mètre cube), inconstante et non maitrisable. Cet élément d’entrée impose des processus progressifs de réduction de la bio-charge qui soient robustes, capables de réduire un large spectre de micro-organismes en quantité de départ très importantes.
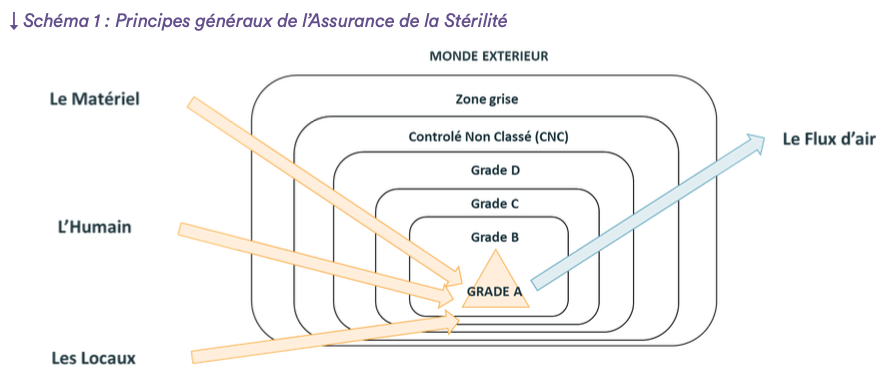
3. Les 4 éléments de protection du sanctuaire
Afin de protéger le sanctuaire du Grade A du monde extérieur “hostile” (en termes de charge microbiologique et particulaire), la plupart des sites de production stérile présentent différentes “couches” de protection telles que représenté dans le schéma 1.
Toutes les couches ne sont pas toujours présentes. Deux exemples typiques sont les suivants :
- L’absence de zone CNC dans certaines usines : dans ce cas, la transition entre la zone “grise” et le Grade D devra faire l’objet d’une attention très particulière
- L’utilisation d’un isolateur permet de s’affranchir du Grade B : en conséquence, les phases ou l’isolateur est ouvert (e.g. montage) et où donc le sanctuaire du Grade A est exposé au Grade C impliquent des précautions et protections supplémentaires.
Nous allons maintenant détailler les différentes mesures qui doivent être prises autour de 4 éléments principaux (le matériel, le personnel, les locaux, les flux d’air) dans le cadre de la fabrication d’un médicament stérile.
3.1 Le matériel
Les processus de réduction de la bio-charge vont depuis la stérilisation par la vapeur (un des processus les plus robustes) jusqu’à la désinfection manuelle (processus le plus fragile). La nature chimique et/ou physique de la désinfection ou de la stérilisation est un élément important mais les aspects de mise en œuvre (automatique ou manuel) le sont également. L’analyse des deux éléments (système de réduction de la bio-charge et mise en œuvre) pris de manière holistiques doit permettre d’établir la stratégie la plus adaptée aux process et à son design et d’identifier les risques résiduels.
Les processus de réduction de la bio-charge se font en général à l’interface entre deux niveaux de propreté différents afin de nettoyer et de diminuer la bio-charge sur les matériels ou les équipements. Cela se fait par exemple avec des autoclaves entre des zones de classe C et des zones de classe B. Cela peut également être le cas pour de la bio-décontamination par peroxyde d’hydrogène en phase gazeuse entre classe C et classe B.
Il existe des pratiques avec réduction de la bio-charge par désinfection du matériel dans les sas matériel. Si la désinfection se fait de manière manuelle alors elle est sujette à la variabilité humaine qui est difficilement contrôlable et donc difficilement validable. Il y a là un risque de contamination qu’il est difficile de quantifier. C’est probablement un des risques principaux si la charge microbiologique entrante augmente pour différentes raisons comme évoqué plus loin.
Il est également possible d’avoir un matériel déjà stérilisé et suremballé afin de pouvoir le transporter vers des classes plus propres en retirant une couche d’emballage au passage vers une classe de niveau de propreté supérieure.
Ces processus de réduction de la bio-charge ou de “containment” sont critiques et doivent faire l’objet d’une analyse poussée avec identification des risques et bénéfices associées aux différentes options possibles.
3.2 L’humain
S’il n’est pas possible de stériliser ou de désinfecter, alors il faut emballer l’objet pour contenir les contaminants dans son “emballage”. C’est, par exemple, ce qui est fait pour le personnel entrant dans les zones à atmosphère contrôlée (ZAC).
Le personnel arrivant dans l’usine retire ses habits de ville pour revêtir une tenue usine. Cette tenue a pour objectif de limiter les contaminations apportées par les vêtements qui ont traversé l’extérieur de l’usine. De plus en plus d’usines possèdent une zone propre non contrôlée (CNC) qui protège le grade D de l’intérieur de l’usine. C’est une première barrière où le personnel ajoute une protection comme par exemple, une coiffe, un cache-barbe, une blouse et des surchausses. A partir de cette zone CNC, le personnel entre via des sas dans les zones classées de propreté croissante et à chaque passage de barrière (sas entre deux classes), des protections supplémentaires sont portées pour finalement totalement “envelopper” l’opérateur.
La dernière révision de l’Annexe 1 ne permet plus l’entrée de personnel dans le sanctuaire aseptique (grade A) car le risque de contamination est trop grand. Pourtant, il existe encore de nombreuses situations où le design des installations imposent la présence d’opérateurs en grade A, en particulier pour les produits lyophilisés où il faut transporter les flacons partiellement fermés depuis la ligne de remplissage vers les lyophilisateurs. Sachant que l’humain est la première source de contamination, et même si tout est mis en œuvre pour contenir au mieux les germes humains (habillage) cela reste fragile. Il est donc presque “normal” de trouver un certain taux de résultats positifs en grade A si du personnel travaillent dans ces zones. Nous avons rencontré de nombreuses situations de ce type et lorsqu’une contamination est retrouvée une investigation est initiée alors que la probabilité de trouver une cause spéciale est quasi nulle puisqu’il s’agit ici d’une cause commune au design de l’installation.
Les technologies barrières permettent de protéger le produit et plus généralement les surfaces critiques. L’Annexe 1 va dans ce sens et privilégie ces technologies barrières. Il est aujourd’hui interdit de construire de nouvelles unités aseptiques en utilisant les ancestrales zones aseptiques A dans B.
Si les technologies barrières offrent des avantages certains et diminuent les risques de contamination, il existe des risques propres qu’il convient d’analyser. En particulier, les questions relatives aux transferts de matériels dans un isolateur doivent également être étudiées avec soin. C’est une route de contamination potentiellement massive peu étudiée car les technologies barrières et l’isolateur en particulier donnent un faux sens de sécurité. Toutes les précautions nécessaires dans les processus de réduction de la bio-charge des matériels sont applicables aux technologie barrières comme aux technologies classiques.
3.3 Les locaux
Quel que soit le processus de réduction de la bio-charge, il faut un système de barrières entre l’extérieur de l’usine et le sanctuaire aseptique. Ces barrières doivent bloquer les contaminations et permettre le maintien d’un environnement progressivement de plus en plus propre pour finalement arriver au statut aseptique du grade A.
Les entrées et les sorties entre deux zones de propreté différentes se font par des sas pour le personnel d’une part et pour le matériel d’autre part.
Les sas permettent d’isoler les zones entre elles lors de l’ouverture des portes. Quand une porte est ouverte coté “sale”, alors la porte coté “propre” doit être fermée. Le sas est alors devenu “sale” et il faut un certain temps pour retrouver le niveau de propreté “propre”. Ce temps est appelé “recovery time” bien défini dans les normes ISO 14644 et dans l’Annexe 1.
Au fur et à mesure que l’on se rapproche du sanctuaire de Grade A, le niveau de nettoyage et de désinfection des salles augmente en fréquence et en intensité.
3.4 Les flux d’air
Les barrières sont gérées par les flux d’air qui “poussent” les contaminants des zones le plus propres vers les zones moins propres. C’est le principe du piston d’air et des cascades de pression entre classes de propreté différentes. Pour assurer et maintenir ce flux d’air, une différence de pression d’un minimum de 10 Pascal doit exister entre deux classes particulaires. Ce flux permet également d’évacuer l’air vers les zones les moins propres lors de l’ouverture des portes des sas.
4. De l’analyse au cas par cas vers une veritable approche holistique
On voit là que ces notions simples peuvent trouver des déclinaisons multiples. A chacune de ces déclinaisons, de nouveaux risques peuvent apparaitre. Comme nous l’avons évoqué, un des risques principaux est représenté par les actions manuelles car elles sont, par nature, dépendantes de l’humain. La reproductibilité ne peut donc pas être démontrée.
Quand les grands principes de la réduction de la bio-charge d’une part et du transfert d’une zone de propreté à une zone de propreté supérieure d’autre part sont établis, alors la validation (EMPQ, APS) peut avoir lieu. Les résultats de la surveillance microbiologique EM permettent alors la vérification que l’exécution dans le design défini produit les résultats attendus et en particulier zéro contamination en grade A.
Dans la grande majorité des cas, c’est bien une absence de contamination qui est attendue et observée. Quand un résultat est différent de zéro, il y a le plus souvent une investigation sur le lieu de la contamination et au moment de la contamination car une recherche de causalité dans l’espace et le temps est la première réaction.
Compte tenu de la complexité des processus intimes mis en œuvre dans la réduction de la bio-charge et du transfert de classe en classe, il est plus judicieux de regarder le résultat dans un environnement plus large. Le résultat positif doit être interprété comme un signal d’alerte sur une fragilité potentielle du système dans son ensemble, pas sur la contamination ponctuelle en elle-même.
La question devrait être : qu’est ce qui peut dysfonctionner dans le dispositif en place en termes de design et/ou d’exécution qui ne permet pas une protection suffisante du sanctuaire du Grade A ?
Le signal lancé par un résultat différent de zéro n’est pas forcément lié à un évènement survenu dans l’enceinte de l’usine.
Par exemple si la bio-charge à l’extérieur de l’usine augmente significativement, le système en place peut ne plus être suffisant pour contenir cette augmentation de la bio-charge. C’est une situation typique si des travaux de construction se font à proximité de l’usine ou du bâtiment concerné en raison de l’augmentation de germes de l’environnement souvent plus résistants aux désinfectants classiques. C’est aussi le cas par exemple à l’automne avec une augmentation des spores de champignons qui sont aéroportées.
En réalité, seul un regard holistique au travers d’une analyse systémique peut permettre de trouver les failles du système. C’est ainsi que les causes les plus probables d’une contamination détectée en grade A peuvent trouver des réponses bien en amont du lieu de la contamination. La notion de Contamination Control Strategy (CCS) introduite par la dernière révision de l’Annexe 1 reflète parfaitement cette approche car elle incite les industriels à mettre en place de manière systémique un ensemble de mesures pour protéger le sanctuaire du Grade A.
Conclusion
Cette vision holistique de la maitrise de la contamination repose sur les hommes et les femmes du site de production. Que ce soit dans la définition des approches et des stratégies que dans leur mise en œuvre c’est par les hommes et les femmes que le succès est possible. Les meilleurs processus de réduction de la bio-charge, les meilleures barrières entre classe restent inopérantes sans des personnes compétentes et engagées.
C’est par l’éducation, comprendre le pourquoi des requis, que les principes de l’assurance de la stérilité trouvent des déclinaisons concrètes pour la maitrise de la contamination. Chaque acteur doit devenir le gardien du sanctuaire et cette posture doit s’inscrire dans la culture du site et de l’entreprise tout entière.
Les industries pharmaceutiques qui ont intégré l’assurance de la stérilité dans leur programme d’éducation et dans leur culture ont un avantage compétitif certain car plus à l’abri des observations réglementaires tout en augmentant la qualité des produits et la performance globale.
Antoine AKAR
Yves MOINARD
Partager l’article

