


Sommaire
- Point de vue de la direction de l’inspection (DI) de l’ANSM sur le document ICH Q12
- ICH Q12 : les fondamentaux. Retours des travaux du GIC A3P ICH Q12
- First steps towards ICH Q12: Leveraging process understanding & development data to define process Established Conditions
- ICHQ12 Implementation from an Industry Perspective with a Focus on Established Conditions
- ICH Q12 compliance and Unified Quality and Regulatory Information Management
- Burkholderia cepacia a encore frappé
- Nouveau guideline Stérilisation de l’EMA
- How to store highly sensitive drugs? The benefit of functional coatings
ICHQ12 Implementation from an Industry Perspective with a Focus on Established Conditions
The implementation of the ICH Q12 guideline requires an updated regulatory framework within the industry with the inclusion of Established Conditions related information in regulatory submissions. This is expected to facilitate an improved operational and regulatory flexibility upon its implementation.

This increased flexibility, expected to be achieved through leveraging effective quality systems, knowledge and risk management, will enable both the industry and regulators to bring benefits to patients by reducing drug shortages, support continuous improvement and facilitate innovation.
Special emphasis is placed in this article on the applications of Established Conditions (ECs) concepts for vaccines with specific examples which may be of potentially wider interest to the pharmaceutical industry. These are presented in the context of stimulating, and as an outcome of, ongoing industry discussions and reflections as presented recently at both PDA Biopharmaceutical Conference (September 3-4, Munich) and A3P ICHQ12 (September 19, Lyon) events.
1. Background
The concepts outlined in the ICH Q12 (draft) guidance are a natural evolution and an integral part of the opportunities afforded by the technical and scientific progress made over the last decades through the emergence of the science and risk-based approaches used in drug development (i.e., ICH Quality Guidelines Q8, Q9, Q10 and Q11 on the Chemistry, Manufacturing and Controls (CMC) aspects). Currently, there are limitations in applying more flexible regulatory approaches to post-approval CMC changes as described in ICH Q8 (R2) and Q10 Annex 1; however, these would be made possible for the commercial phase of the product lifecycle, by applying the appropriate and complementary concepts of ICH Q12. 1
ICH Q12 provides a flexible (optional) framework to facilitate the management of post-approval CMC changes across the product lifecycle, in a more predictable and efficient manner, grounded on Quality by Design (QbD) principles and product and process understanding (ICH Q8 and Q11). This flexibility (expected to lead to an enhanced industry’s ability to manage implementation of CMC changes more effectively under the company’s Pharmaceutical Quality System, PQS) is predicated on the effective implementation of the ICH Q12’ tools (e.g., Established Conditions) and enablers (e.g., an effective PQS). Although the application of this guidance is optional, its tools and enablers are linked and complementary to each other as they are working together to provide different degrees of operational and regulatory flexibility.
ICH Q12, introduces the term ‘Established Conditions (ECs)’ for the first time in ICH guidance’s. EC is a term used for ‘approved matters’, colloquially referred to as ‘registered details’ for many years. It should be noted however that ECs have always been mandatory in a marketing application, even though they may not have been called ECs to date. According to ICHQ12, the “Established Conditions”, resulting from development efforts and reflected in an appropriate control strategy, are the elements in an application that “are considered necessary to assure product quality and therefore would require a regulatory submission if changed post-approval”.
According to the current draft Guideline, examples of established conditions include the critical process parameters, critical material attributes, elements of analytical procedures assuring proper performance as well as generally understood critical elements like drug substance name and structure, manufacturing sites, specifications, storage conditions and shelf-life, etc.
The extent of regulatory and operational flexibility which can be achieved upon ICH Q12 implementation is therefore linked to the proper identification and justification of the Established Conditions, and to the consequent agreed and predicted (as referenced in the Product Lifecyle Management PLCM document) change management strategy, applying risk management principles (ICH Q9), and a robust pharmaceutical quality system.
An industry perspective on ICH Q12 implementation, with special focus on Established Conditions with illustrative examples for Biologics (focus on vaccines), is outlined here.
2. Discussion and Examples
Vaccines are complex biological products, as they may be composed of one or more antigens in combination, with unique molecular structures presented in a final formulation potentially with an adjuvant system of varying composition. Vaccines are also characterized by very long manufacturing and/or product life cycles (e.g. Measles-Mumps-Rubella, Hepatitis recombinant B vaccines). Many of today’s vaccines were registered decades ago but are still produced and used to immunize millions of people. Altogether, this means that vaccines licenses require submission of multiple variations or supplemental applications over an extensive number of years and, in addition, because they are often combination of multiple antigens or use the same adjuvant system, changes are often repeated across multiple product licenses and jurisdictions (often with divergent risk categorization of changes), leading to a highly complex and challenging management of their life-cycle and supply availability.
Thus, to enable an effective lifecycle management that would also appropriately mitigate vaccine’s supply availability risks, the identification of Established Conditions (ECs) is a prerequisite for the ICH Q12 deployment strategy. This would be based on a pro-active structured, standardized approach to Post-Approval Changes to these ECs, where the future EC change and its reporting category would already be determined. This information would be reflected in the Post-Approval Change Management Protocols (PACMP) and PLCM documents. These pre-approved registered details would then be legally binding.
2.1 EC Application
The application of EC is by default easier for new medicinal products which have been developed based on a QbD structured, enhanced approach following ICH Q8–Q11 guidelines. For registration of new products there is already a proactive planning and risk evaluation of potential post-approval changes included in the regulatory filing and without any pre-approved “registered details”.
In contrast, for commercial products that might have been developed using the traditional approach, the application of EC could be based on a limited understanding, for example based on retrospective data analysis and rationale. Due to the limited knowledge of the relationship between inputs and resulting quality attributes which could lead to a potentially large number of inputs and outputs, there could be a higher number of EC thus limiting the flexibility afforded by the application of ICHQ12. Hence, a stepwise approach and implementation of ICH Q12 where value added (business and technical perspective) might be preferred.
Regulator feedback indicates that ICHQ12 expectations for commercial assets include building on the current data set and knowledge where appropriate through a retrospective data analysis without the need to redevelop these products. Additionally, the application of Q12 guideline, requires a change in current (mostly reactive) life-cycle management mindset, essentially along three axes: (1) a review of existing regulations, (2) consideration of approved details, and (3) pro-active planning and risk evaluation of proposed ECs changes together with the associated data generation and validation or comparability strategy.
All marketing applications contain a combination of ECs and supportive information; supportive information is not considered to be an EC. More details about the CTD sections that contain ECs are presented in Appendix 1 of the ICH Q12 guideline.
The identification of ECs provides an opportunity for the industry to pinpoint exactly the critical elements within the overall information provided in the CMC part of a Marketing Authorization Application (MAA), and consequently, to focus dossier maintenance on these only and not on the maintenance of other type of information (i.e., “supportive” information).
ECs are to be identified (with appropriate justification) in Marketing Applications in all ICH countries’ dossiers within specific Common Technical Document (CTD) modules. The PLCM document will lists all the ECs and their change reporting categories. Obviously, if ECs are not identified in the Marketing Application or in specific CTD modules, then normal regulatory post-approval change management processes will apply in these situations.
Among the ECs specified in the Appendix 1 of ICHQ12 some are unambiguous and therefore will not be discussed below, for example the drug substance names, structure, manufacturing sites, shelf-life, etc. Special care and consideration should be taken when defining and identifying the ECs in the manufacturing and analytical sections of the dossier (including analytical procedures, specifications and reference standards and materials) as these will frequently vary during the product lifecycle according to the company development approaches, product and process and analytical understanding and the potential risk to product quality as it relates to patient safety and efficacy.
Due to the existence of a science and risk-based approach and to a well-defined regulatory framework (ICH Q8, Q9, Q10 and Q11), manufacturing (especially process parameters) was identified as a priority area of ECs identification. In contrast, for analytical procedures, reflection is still ongoing on the application of the ECs concept, with a close view on the upcoming ICH framework through Q2 Validation of Analytical Procedures revision and new Q14 Analytical Procedure Development.
2.2 Manufacturing
The use of an enhanced approach to identifying ECs for the manufacturing process should build on current elements of the control strategy, which themselves are based on the science and risk-based approaches mentioned above. In addition to the unit operation and the sequence of steps, and considering the overall control strategy, ECs in a manufacturing process description (3.2.S.2.2/P.3.3 CTD sections) should be those inputs (e.g., process parameters, material attributes) and outputs (e.g. in-process controls) that are necessary to ensure product quality.
This EC identification requires an understanding of interaction between inputs and product quality, safety and efficacy attributes (QbD processes like risk and Critical Process Parameters (CPP) assessments) together with a corresponding control (including testing) strategy (ICH Q8-11).
It is also made clear in the guidance that, irrespective of the approach used to identify ECs, a suitably detailed description of the manufacturing process is still important to provide a clear understanding of
how the process is being run and that the use of this guidance should not lead to a less detailed description of the manufacturing process in Module 3 CTD dossier of the Marketing Application.
Note that the examples presented in this article were developed as part of industry efforts (Vaccines Europe) to prepare for the upcoming ICH Q12.
An example of the application of EC for a vaccine’s formulation process step with the adsorption of an antigen on aluminium is outlined below in Figure 1.
A major Critical Quality Attributes (CQA) for that formulation step is the completeness of adsorption that could be controlled through release and stability testing. During that step, the pH operation target has been identified as a critical process parameter (CPP); it is well controlled, monitored and adjusted on-line. Following the enhanced development practice, this parameter has been extensively studied across a realistic range during the process development in combination with other parameters that were identified as having an influence. Through this development work it has been identified that pH has a strong influence on the CQA completeness of adsorption. The pH operation target therefore is considered as an EC. As the knowledge of the impact/risk of change to the EC on the CQA within a reasonable range is well understood, and the parameter is well controlled, any change within the operating range studied would have a low-quality impact, thus justifying a Notification Reporting Category (in the absence of the EC approach, this change would be considered as a prior-approval reporting category, as this is a change to the manufacturing registered detail).
Note that, if based on knowledge, the EC for pH parameter would have been defined as the pH operating range, rather than the pH operating target as described above, any change of the pH operating target within the registered (EC) operating range would not require any regulatory action as there would be no impact on the product quality, safety and efficacy. However, a change outside of the range would require the pre-determined classification of action. Given that the CQA completeness of adsorption is tested in each batch, pH and other influencing parameters are also under good control within minimal process variation, then we would be looking to prospectively justify any such change as a Notification.

2.3 Analytical (Procedures and Specification acceptance criteria)
Similarly, the identification of ECs in the analytical sections of the Dossier, should build on already established elements of the control strategy (analytical controls’ understanding). For example, on analytical QbD like critical method parameters (CMP) assessment, testing strategy and specifications, and on parts relating to analytical procedures which are currently under discussion and considerations in the upcoming ICHQ14 and ICHQ2 revision.
As specified in the Q12 guidance, ECs related to analytical procedures should include elements which ensure performance of the procedure. Appropriate justification should be provided to support the identification of ECs for analytical procedures. Although the extent of ECs could vary based on the method complexity, development and control approaches, an appropriate detailed description (with both ECs and non-ECs) of the analytical procedures in Module 3 CTD sections (3.2.S.4.2, P.4.2 and P.5.2) of the MAA is still expected to provide a clear understanding regardless of the approach used to identify ECs for analytical procedures.
Two examples of the application of EC in the analytical area are outlined below related to both specifications and reference standards.
As can be seen in Appendix 1 of the ICH Q12 guidance, the specifications described in CTD sections 3.2.S.4.1, P.4.1 and P.5.1 are expected to be ECs as they are part of the overall controls ensuring product quality. However, care and considerations should be taken when identifying ECs for the specification attributes, as we might also encounter non-ECs (with appropriate justification) within the specifications, as exemplified below in Figure 2 for the application of EC for specifications of glycoconjugate vaccines.
Glycoconjugate vaccines are synthesized upon reaction between a carrier protein and a poly-dispersed mixture of poly- or oligo-saccharides (antigens). After this conjugation step, residual, unreacted carrier protein may be present in the purified drug substance, depending on the conjugation reaction and/ or purification steps efficiency.
The residual (free) carrier protein is not considered as a Critical Quality Attribute, as it has no impact on safety and efficacy, but may be included in the specifications panel (release) to support verification of manufacturing consistency, if needed. Thus, the free carrier protein is not an EC as it is not directly related to safety and efficacy of the product, although being included in the specifications. Changes to the residual (free) carrier protein test should be therefore managed internally within the company PQS, without prior approval from or notification to the Health Authority, provided that the change does not impact product quality, safety and efficacy.
This example illustrates the difficulties that might be encountered when applying the EC concept of ICHQ12 without adaptation to the current regulatory framework and expectations as non-CQA specification attributes can often be encountered in other medicinal products. According to the current regulatory framework and expectation, in the absence of the EC approach, the whole specification would be registered detail, regardless of the assessment of impact to their safety and efficacy and as such any changes would require prior-approval. Similarly, the current FDA thinking according to the draft guidance (Chemistry, Manufacturing, and Controls Changes to an Approved Application: Certain Biological Products, of Dec 2017) would require a prior-approval supplemental application for changes to release specification. In addition, specifications are part of the Lot Release Protocol and therefore need to be communicated. This is further elaborated under the Challenges associated with the application of EC concepts.

Another example of the application of EC for reference standards is outlined below in Figure 3.
A reference standard (RS) is a material used in the release or stability testing of batches of material or product. Both chemical and biological standards can be used as reference standards. Due to the biological nature of some vaccines reference standards, RS qualification occurs frequently throughout the life cycle of the product, at either stock-out, or at expiry.
A change in RS (batch) often follows an agreed qualification protocol (i.e., QP and CP protocols today, similar in many aspects to PACMP in ICHQ12) where the type of qualification studies to be performed and assessment criteria are specified. The approach and level of detail given in justifying ECs for a vaccines assay or in a qualification protocol is a company decision and may depend upon other elements associated with the overall control of the vaccine. Simple approaches may be acceptable to regulatory authorities when the risks to patients are minimal.
As proposed in Appendix 1 of the ICH Q12 guidance, in CTD sections 3.2.S.5 and P.6, the EC corresponds to the specifications (i.e., qualification acceptance criteria demonstrating that a given reference standard is fit for purpose) of the Reference standard.
Thus, there is no need for regulatory action if there is no change to the ECs for the implementation of a new Reference standard, if its specification (i.e., EC) does not change. The change is managed under the PQS, according to a qualification (i.e., PACMP type in ICHQ12) protocol as described in the original license or in subsequent variations. Currently, in the absence of the EC approach, for example a change to the current reference standard details like its batch number would be considered, in some jurisdictions, as a prior-approval reporting category, as a change to registered detail.

2.4 Challenges associated with the application of EC concepts
There are potential challenges associated with the application of the EC concepts of ICH Q12 due to regulatory environment & framework and manufacturer’s considerations as outlined below:
A. External regulatory environment and framework:
• There is a risk of regional nuances in Q12 implementation and of existing regulations (expectations or precedents) as in certain ICH regions, the current ICH Q12 guideline is not fully compatible with the current established, or proposed, legal framework (e.g., EU variation Guidance, the draft FDA guidance Chemistry, Manufacturing, and Controls Changes to an Approved Application: Certain Biological Products, of Dec 2017). Therefore, for implementation of Q12, a change in regulations may be needed in some regions.
• The regulators expectations in the adoption of ICH Q12 needs further definition. For example, an understanding of the acceptability of adoption of its elements in a step wise approach versus all or none (i.e. can elements of Q12 such as EC, PACMP, PLCM, be implemented in stages or must it be done all at once).
• The requirements for intra- or inter- companies’ consistency in EC identification and reporting should be clarified (i.e. what level of harmonization of the approach to EC is expected).
• Additionally, the requirements for the maintenance of PLCM document where all the ECs are listed, needs to be clarified.
B. Other challenges are linked to the manufacturer considerations:
• The limited enhanced (QbD) knowledge for some commercial products increases the possibility of identifying a high number of ECs because of limited risk-based knowledge. This may then require either the generation of new data to justify PACs under Q12 or, more effectively, the leveraging of existing manufacturing data and extrapolate this knowledge for criticality and risk assessments.
• Converting existing “registered” information and regulatory mechanisms to ECs (i.e., how to reflect in our dossiers the updated knowledge on the existing supporting information (some of it seen today as “binding”) if now determined as non-EC type information).
• It is also necessary to consider the potential complexity of the need to manage PAC if ECs concepts are not applied for all products.
• There is also the risk of having different ECs registered worldwide because of disparity of regulatory authorities’ acceptance or implementation of ICHQ12 concepts.
• The value added in the conversion of some of our commercial products dossiers to ICH Q12 assets, as this activity would need to be evaluated and prioritized based on defined criteria.
Despite the challenges listed above, ICH Q12 offers opportunities to maintain the regulatory oversight while at the same time leveraging effective quality systems, knowledge and risk management at manufacturer side, thus enabling operational flexibility. This would then bring benefits to our patients by reducing drug shortages, support continuous improvement and facilitate innovation.
2.5 Post-Approval Changes to EC’s
As shown by the above examples, it is important to properly identify and justify the ECs in the applicable CTD sections of the regulatory applications and to propose a reporting category if they are changed because as per ICH Q10, companies that apply the principles and concepts of ICH Q8, Q9 and /or Q10 should be eligible for reduced regulatory oversight when they demonstrate that an effective PQS is in place.
Thus, with sufficient product and process understanding and the use of Quality Risk Management, certain post-approval changes should be managed in the PQS only or as a regulatory notification (with no or limited prior approval by regulators) when a comprehensive risk assessment concludes that a proposed change introduces no risk to patient safety, product quality and efficacy. In other words, companies can manage more changes without the need for prior regulatory approval, provided they operate under a framework including an effective PQS, along with sound product and process knowledge and risk management practices.
ICH Q12 complements and adds to the flexible regulatory approach to PACs described in Q10 Annex 1 by introducing a clear framework encompassing a risk-based categorization of Post-Approval CMC Changes linked to the potential impact on the overall product quality of the product, as it relates to patient safety and product efficacy; the level of reporting being commensurate to the level of risk:
• Prior-approval when high risk to product quality
• Notification when there is a moderate to low risk to product quality
• Internal management within the company PQS with no regulatory reporting for changes with no or lowest risk to product quality.
Hence, an effective PQS as described in ICH Q10 and in compliance with regional GMP requirements where the application is filed, is necessary across the entire supply chain and product lifecycle to support the use of the ICH Q12 tools.
According to the ICH Q12 tools and enablers, all the ECs (including their regulatory reporting category if changed, see ICHQ12 Figure 1 (i.e., Decision Tree for identification of ECs…)) are to be agreed and listed in the PLCM document and their changes reported accordingly (i.e., changes to ECs are expected to be reported to Regulatory Authorities at least via notification).
2.6 Standardized Post-Approval Change Management
The extent of operational and regulatory flexibility is subject to product and process understanding (ICH Q8 and Q11), application of quality risk management principles (ICH Q9), and an effective pharmaceutical quality system (ICH Q10), all enabled by an appropriate knowledge management.
Senior Quality Leaders of the pharmaceutical industry are working together within the ‘One Voice of Quality’ initiative coordinated by the PDA (Parenteral Drug Association) to establish industry standards for a consistent and robust risk-based management of PACs and to define the set elements required to demonstrate the effectiveness of the PQS linked to Change Management.
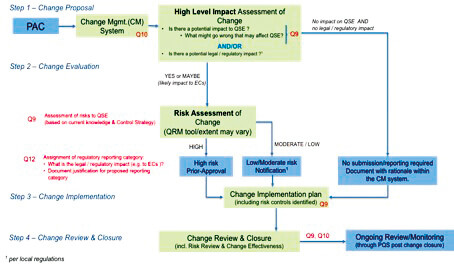
The (draft) decision tree related to the Risk-based Assessment of PACs, to be published soon as part of a document provisionally entitled ‘Effective Management of Post Approval Changes in the Pharmaceutical Quality System (PQS) – Through Enhanced Science and Risk-Based Approaches’, is presented in Figure 4 below.
These efforts are made to build trust with regulatory agencies so that low risk changes can be managed within the PQS or to be reported to Regulatory Authorities via notification, thus reducing the burden (for both industry and regulators) associated with the management of PACs.
Conclusion
Both regulators and industry should focus on what really matters, which is the timely access to safe and efficacious medicines for patients. This is aligned with the stated objective of the ICH Q12 guideline “A harmonised approach regarding technical and regulatory considerations for lifecycle management will benefit patients, industry, and regulatory authorities by promoting innovation and continual improvement in the biopharmaceutical sector, strengthening quality assurance and improving supply of medicinal products.”
The implementation of ICHQ12 requires a framework within the industry which builds on prior knowledge, science and innovation to identify ECs with a structured approach to product development and lifecycle. This updated framework is crucial for any accelerated development of new products but is posing some additional challenges to its application for commercial products.
The use of Q12 tools and enablers will facilitate a more effective PACs management during lifecycle by providing a risk-based approach to define level of regulatory oversight and by anticipating future changes and their reporting categories. Additionally, effective PQS, Knowledge and Risk Management can be leveraged effectively to enhance operational flexibility, secure supply continuity and foster innovation.
As this new ICH guideline is not yet adopted (at Step 2b) we will have to wait and see if these current considerations, tools and enablers of ICH Q12 will remain unchanged when reaching Steps 4 and 5. Further finetuning of the above EC approach may be required in line also with (as needed) regional regulatory framework updates due to this guidance implementation, hopefully worldwide and not only in ICH regions. Although ICHQ12 guidance has now been ICH adopted at Step 4 on Nov 20 2019, the concepts and details described in this article remain valid.
Partager l’article

Glossary
ICH: International Council for Harmonisation
EC: Established Conditions
PACMP: Post Approval Change Management Protocol
PLCM: Product Lifecycle Management
CMC: Chemistry, Manufacturing and Controls
QbD: Quality by Design
QRM: Quality Risk Management
QSE: Quality, Safety, Efficacy
CQA: Critical Quality Attributes
CPP: Critical Process Parameters
CMP: Critical Method Parameters
CP/QP: Comparability (USA terminology) and/or Qualification (EU terminology) protocols
PAC: Post-Approval Changes
MAA: Marketing Authorisation Application (EU terminology)
MA: Marketing Applications used here to denote regulatory applications and jurisdictions outside EU
PAS: Post Approval Supplement
PQS: Pharmaceutical Quality System
Bibliography
Guidance for Industry: Draft ICH Guideline Q12 on the Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management, Step 2b, 14 Dec 2017, EMA/CHMP/ICH/804273/2017
Mihai Bilanin – GSK VACCINES
Mihai Bilanin (Global Regulatory, CMC Excellence) holds a PhD in Chemistry and has spent over 17 years in the pharmaceutical industry, with the last 15 years in Global Regulatory Affairs in Canada and Europe. He has managed worldwide regulatory projects in development, registration and life-cycle management (various degrees of complexity). Lately, Mihai has focussed on regulatory matters related to the Quality by Design approach and ICHQ12, in addition to managing a team of Regulatory CMC writers.
Siobhan Ahern – GSK VACCINES
Siobhan Ahern (Global Quality, Technical Lifecycle, GSK Vaccines), has an international executive MBA and prior to working at GSK has over 10 years of experience in marketing roles of increasing responsibility at a global provider of polymer materials. She has been at GSK vaccines for over 5 years working in project management, lifecycle and her current role as Technical Lifecycle in global quality.
Marcello Colao – GSK VACCINES
Marcello Colao currently holds the position of Director Regulatory & Technical Lifecycle within the Global Quality organization of GSK Vaccines aimed at sustaining and leveraging the successful lifecycle management of GSK vaccines from both a regulatory and technical perspective. Marcello joined GSK Vaccines in 2012 as Director Quality & Regulatory Compliance to lead the Regulatory Conformance Transformation of the company. Prior to that, he worked for Pfizer where he held positions of increasing responsibilities within the Global Quality Operations organization. Marcello holds a Master Science degree in Chemistry and has 20+ years’ experience in the pharmaceutical industry with an extensive knowledge of pharmaceutical operations, Quality Systems as well as Regulatory Compliance matters.
Cristiana Campa – GSK VACCINES
Cristiana Campa, PhD, is currently a Technical R&D Advisor and Fellow at GSK Vaccines, with 20 years’ experience in biologics and related analytical and development strategies, gained in different universities and companies. She joined Novartis Vaccines in 2006, focusing on development, validation and transfer of analytical methods for release and characterization of several vaccine products, first as senior manager and then as Head of Analytical Development, Italy. Since 2012, Cristiana has worked on Quality by Design (QbD) principles implementation for vaccines. After acquisition of Novartis Vaccines by GSK in 2015, she has been the Head of QbD Integration and, until June 2018, the Head of Science and Development Practices in Technical R&D, covering QbD implementation, Knowledge Management and Development roadmaps.
Geoffroy Geldhof – GSK VACCINES
Geoffroy Geldhof is an Expert scientist at GSK Vaccines Research & Development center in Rixensart, Belgium, where he works in microbial drug substance upstream platform. From 1997 to 2007 he was a scientist at Eli Lilly and company where he developed chemical process for clinical production of API. Since he joined GSK in 2007 he has made contributions in improving adjuvants synthesis and purification, conjugate vaccines, and downstream process. He spent the last three years implementing Quality by Design in the organization. He has an M.S. in chemical engineering from the Meurice Institute, Brussels.
