
Sommaire
- Point de vue de la direction de l’inspection (DI) de l’ANSM sur le document ICH Q12
- ICH Q12 : les fondamentaux. Retours des travaux du GIC A3P ICH Q12
- First steps towards ICH Q12: Leveraging process understanding & development data to define process Established Conditions
- ICHQ12 Implementation from an Industry Perspective with a Focus on Established Conditions
- ICH Q12 compliance and Unified Quality and Regulatory Information Management
- Burkholderia cepacia a encore frappé
- Nouveau guideline Stérilisation de l’EMA
- How to store highly sensitive drugs? The benefit of functional coatings
ICH Q12 compliance and Unified Quality and Regulatory Information Management

1. How ICH Q12 Relates to QMS and RIM?
The implementation of ICH Q12 guideline (currently in draft stage) impacts Pharmaceutical companies’ Quality and Regulatory systems:
• On the Pharmaceutical Quality System (PQS) level, it redefines the process of managing post-approval chemistry, manufacturing, and controls (CMC) changes, impacting the QMS & the ERP
• On the Regulatory level, ICH Q12:
· Impacts the content of pharmaceutical product eCTD submissions
· Changes the process of how to determine which CMC changes require submission or notifications to authorities
· Changes the content of post-approval CMC change-related submissions
Efficient integration between ERP, quality and regulatory management systems will be even more important than before to achieve compliance and attain the potential benefits in product quality.
In this paper, we will examine the different approaches to make these different systems work together and how it relates to a smooth implementation of ICH Q12.
2. The Context of Modern Regulatory Systems
An important part of the current transformation of life science companies is the thorough examination of key Regulatory processes that are critical to getting a product to market quickly and maintaining its regulatory approval.
Processes that span organizational boundaries are especially significant as they are often made more complex by disconnected IT systems and compartmentalized operating procedures. One process that is particularly important is the process of managing variation submissions – mostly due to their time sensitivity and regulatory impact.
CMC change-related submissions are one type of variation submission process. We will identify areas of risk and opportunities and show how to mitigate those risks through unified Quality and Regulatory Information Management.
3. Managing Variation Submissions
The efficient management of variation submissions can be challenging. While these submissions are vital to keeping approved products on the market, the essential processes and technologies to effectively manage them are often overlooked.
The work required to fully implement a manufacturing change and create its accompanying variation submission is typically performed by different departments within a company – Manufacturing, Quality Assurance (QA), Supply Chain and Regulatory Affairs.
Often, each team completes their work separately with little or no visibility into the activities being performed by the other teams. To make matters worse, the teams may lack a comprehensive understanding of the overall variation process and how their work impacts that of the other departments and the company as a whole.
Besides, the outsourcing of some or all of the process can further complicate the situation.
This combination has the potential of introducing the risk of a product non-compliance. To mitigate this risk, many companies have initiated efforts to harmonize processes and standardize the interactions between contributing teams with the intent of increasing efficiency, improving communication and eliminating bottlenecks.
The result of these initiatives is typically a catalog of process maps, RACI matrices and standard operating procedures (SOPs) each intended to build consensus among stakeholders, recognize breakage points and identify areas for improvement.
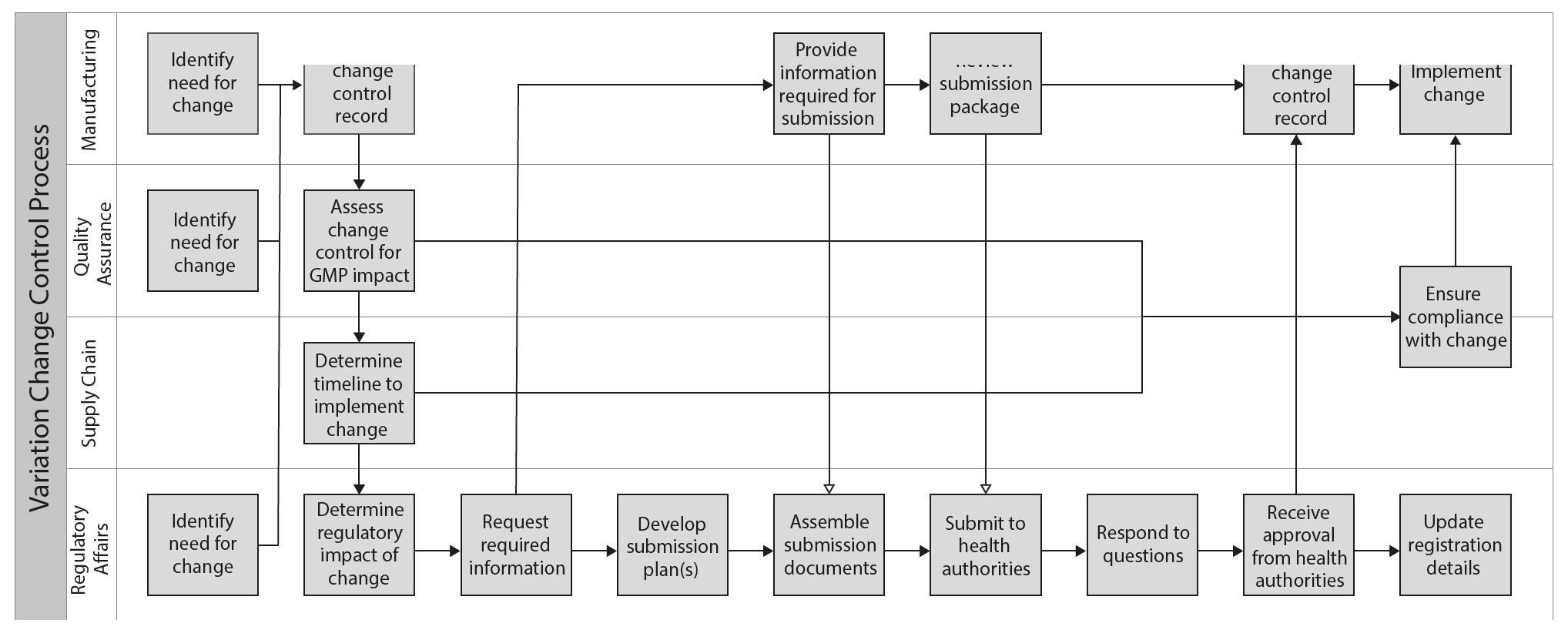
Let’s consider a very basic process map to illustrate the organizational and technological interdependencies associated with a variation change control and submission.
The process map shows each team’s responsibilities and the required interactions between them – answering all of the “what” questions:
• The verbs “create”, “assess”, “request” and “develop” are clear calls to action and the swim lanes establish accountability for each step in the process.
• The linear progression of the process steps implies answers to the “when” questions but provides little or no guidance on specific activity timings, lead-times or durations.
What is missing from this recipe altogether are the answers to the “how” questions:
• For example, when Regulatory Affairs requests information from Manufacturing to support the submission is this done through email, by telephone or using some other means?
• When Manufacturing reviews the submission package prior to dispatch to the health authority is it performed on paper or electronically?
• Finally, how is Manufacturing informed of health authority approval so the change can be implemented without delay?
Now, more than ever before, companies are relying on enterprise information systems to help them answer these “how” questions – relying on each system’s functionality to enforce compliance with their processes and procedures.
However, systems that are disconnected or loosely integrated can actually complicate matters, adding unnecessary overhead, introducing compliance risk and making it more difficult to meet the aggressive timelines required to prepare and publish compliant regulatory submissions.
4. The QMS – Critical for Reliable Quality Management
The Quality Process Management Challenge: Regulated companies must control their Quality processes (Deviations, NCs, CAPAS, CCs, Audits…) for regulatory compliance and to make them as efficient and effective as possible. But manual, paper-based processes are:
• Hard to manage (require a lot of manual work)
• Difficult to track (processes can get “forgotten” for some time)
• Difficult to control and to make reporting on
• Slow
• Error prone at all stages of the process
• Difficult to scale and deploy globally
The eQMS: The solution is to implement an electronic Quality Management System (eQMS or QMS). An eQMS enables to model and execute each quality process as an automated workflow. The benefits are numerous:
• Processes are always executed as specified – no more “Ad hoc processes”
• The progress of ongoing processes is always visible. Managers can monitor any workflow, visualize pending and completed tasks and intervene when tasks become delayed.
• Traceability is built in, facilitating compliance and audits
• Each participant in a workflow has immediate access to the information it needs to complete his/her task
• Automatic alerts & email notifications keep processes on schedule
• Processes can be made multi-lingual, facilitating global operations
• Reporting tools enable to automatically generate and follow-up the KPIs of each process
• Quality processes can be integrated with other IT systems and processes.
When a variation change control process occurs, the QMS is the originating system for the change control, and enables to keep track of the progress of the whole change control. It is thus arguably the most critical element needed to smoothly implement ICH Q12.
5. Understanding the Systems Involved
There are five enterprise systems involved in the management of the variation change control process:
• Enterprise Resource Planning (ERP):
· Manages and tracks raw materials, active ingredients and finished products
· Facilitates production planning throughout the manufacturing process
· Provides visibility into stock levels to ensure order fulfillment and on-time delivery
· Ensures quality control requirements and regulatory compliance obligations are met at all times.
• Quality Management System (QMS):
· Automates, centralizes and consolidates the capture, tracking, management and regulatory reporting for all quality processes and practices
· Common processes include the management of deviations, non-conformances, complaints, audits, change controls, and CAPAs.
• Electronic Document Management System (EDMS):
· Manages the lifecycle of digital content files (documents, spreadsheets, images, etc.) from initial creation through archiving
· Provides version control
· Facilitates review and approval cycles and manages the distribution
· Regulates access to content files through rights management
· Provides metadata-based and full-text searching capabilities.
• Regulatory Information Management Systems (RIM):
· Manages and tracks medicinal product details and registration information
· Product details include API, manufacturing, packaging, distribution, composition and stability information
· Plans and tracks regulatory submission projects and related activities
· Manages correspondence and commitments between the sponsor and health authorities.
• Electronic Publishing System (EPS):
· Builds regulatory submission structures
· Publishes and validates compliant submissions in a variety of output formats (CTD, eCTD, NeeS, VNeeS, eCopy and paper)
· Provides bookmarking and hyperlinking to improve navigation
· Accesses and links regulatory content from EDMS or file system.
In many instances, these systems operate independently from one another – each serving the unique needs of their users.
However, much of the information that is managed within each system is common to all of them. The most obvious example of this is the Active Pharmaceutical Ingredient (API).
To illustrate the impact of this data redundancy, let’s consider the case of changing an API manufacturing site. The API is directly linked to :
• The packaged product SKUs managed in the ERP
• The change control record managed in the QMS
• The API manufacturer documents in the EDMS
• The submission sequence for the variation in the EPS system
• The product registration information in the RIM system
Changing the manufacturing site in the ERP system triggers a cascade of updates that must be made in each of the remaining four systems to maintain compliance. If this process is manual, it must be well-coordinated and controlled.
6. System Interaction: Data and Processes
To further appreciate the scope of the data and process dependencies between these systems consider the following diagram:
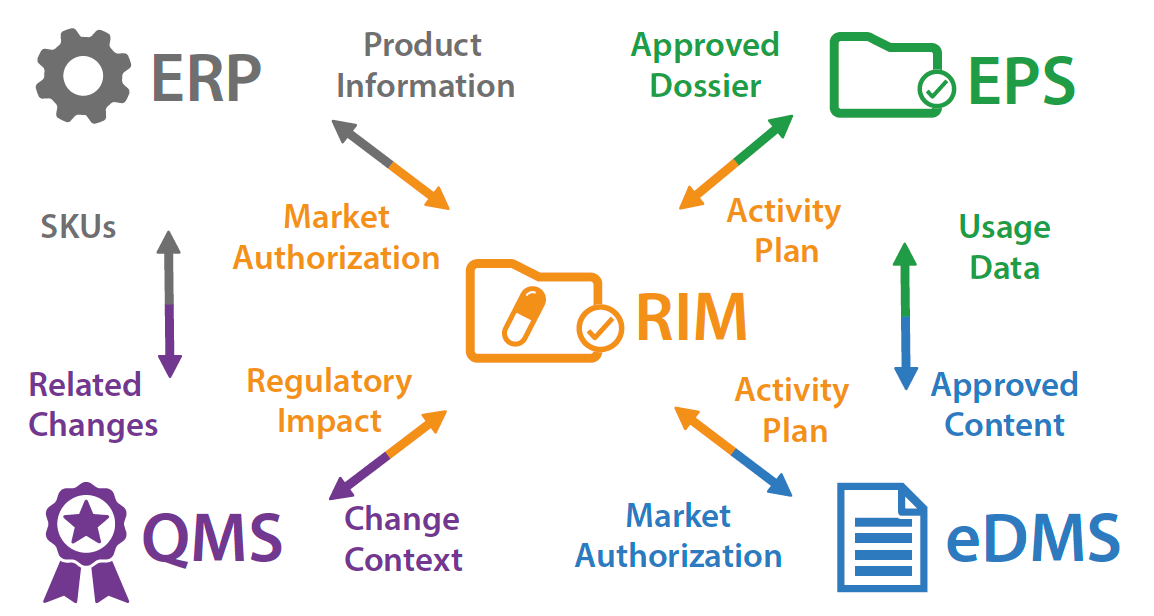
The end-to-end variation change control process requires linkages between the systems to either directly access data (e.g. packaged product SKUs, serialization information) or reference data (e.g. approved submission documents and dossiers).
More specifically, when a post-approval CMC change occurs:
• The QMS manages the change control process
∙ The context of that change is useful for managing activity plans and activities in the RIM
• The ERP manages reference product-related information that might have to be modified following the change
∙ This modified product information must be made available to the RIM which holds the reference regulatory product database (xEVMPD and other information)
• The RIM manages the activity plan and the corresponding regulatory activities necessary to implement that change
∙ The progress of these regulatory activities is useful for managing the change process in the QMS
• The EDMS is used to manage the documents related to the change: SOPs, Risk-based analysis of the regulatory impacts of the change control (based on the ECs & PACMP for ICH Q12 compliant systems), PLCM document, documents that are included in the eCTDs and that will be part of the submission variations
∙ The PACMP (Post-approval change management protocol) document should be available from the corresponding CC process management in the QMS
∙ eCTD Document revisions are useful information for the EPS system, including the revised PLCM (Product Life Cycle Management) document if ICH Q12 is followed
∙ The risk-based impact analysis of the proposed change is useful for the change control process
∙ impacted SOPs can be useful to track from the relevant regulatory activities
• The EPS compiles the eCTD variations to be submitted to the different authorities in each product/country
∙ The approved dossiers should be made available to the corresponding RIM activities
It is easy to understand that one CMC change can trigger a cascade of activities in the RIM (for each product and country involved), a large number of documents to be revised in the EDMS and several variations to be submitted to authorities.
It is easy to understand that one CMC change can trigger a cascade of activities in the RIM (for each product and country involved), a large number of documents to be revised in the EDMS, and several variations to be submitted to authorities.
In the worst case, there are no links. In this case, references to this information are managed manually. This is done by re-keying referenced record IDs or URLs into the other systems or maintaining a list in departmental Excel spreadsheets. These practices, because they rely completely on human action, are inefficient and error-prone which introduces non-compliance risk.
Another way to address the issue are automated links between the systems. Automated links eliminate the need for human intervention and increase the reliability of the referenced data. Automated links can be implemented in one of two ways – interfaces or integrations.
6.1 Interfaces
Interfaces are lightweight links that simply share information between the systems. This information could be a common key such as a marketing authorization number that links to a particular SKU or a change control number that links to a series of variation submissions. Examples of these shared fields are further illustrated in the following diagram:
With a system interface, only data is shared – which does little to ensure the “how” questions are being answered consistently.
6.2 Integrations
System Integration is a more robust approach to system linkage which includes both shared processes as well as shared data.
In the example above, a system integration could cause the RIM system to notify the ERP system which SKUs are authorized for production once regulatory approval is received.
In the case of change control:
• The QMS could trigger a process within RIM to initiate a regulatory assessment of the change and its impact on the specific SKUs covered by the marketing authorizations in each country or region.
• Activity plans for the affected registrations could be created and workflow notifications sent to the Regulatory CMC and Publishing teams requesting new or updated documentation and new eCTD submission sequences.
System integration removes process variability – which is the key to mitigating compliance risk.
However, system integration can be quite complex, difficult to maintain and expensive :
• Often, the systems (particularly the older legacy ones) have different architectures that make integrating with them nearly impossible.
• Updates to one system can have an adverse effect on the integrations with the others, requiring extensive testing and re-validation.
Integrations that were not well designed and have evolved over time are particularly dangerous. In many cases, the original designs were not well documented or the design documentation has not kept up with the evolution of the integration.
This confluence of circumstances often leads companies into a state of “system paralysis” where their manual processes must be maintained in order to compensate for the limitations of their legacy enterprise systems.

7. A Better Way: A Unified Platform
Imagine a solution where:
• All the information required to effectively manage the variation process is available from a single authoritative source.
• Regulatory data, documents, dossiers, and the process workflows to manage those assets are intrinsically linked together facilitating seamless operation and efficient communication across departments.
• Everyone references the same controlled vocabularies.
• Regulatory changes are implemented for everyone, globally, at the same time.
• Closing the loop on a change control once final regulatory approval is received becomes automatic.
• Information from across the value chain can be leveraged to improve organizational performance and achieve operational excellence.
To some, this solution sounds unrealistic and will be dismissed straight away. To others, this solution sounds intriguing and warrants a closer look. And finally, to others still, this solution sounds very familiar because it is what they use every day to achieve world-class status with respect to unified Quality and Regulatory Information Management.
The unified platform is not simply a collection of purpose-built applications (or modules) that are integrated through a series of pre-built connectors.
The unified platform is a highly configurable software architecture that combines:
• A common and engaging user experience (UX)
• Core capabilities: Business process management, document management, data/forms management, business intelligence, traceability
• Unified applications constructed on top of this foundation, using the powerful configuration capability of the architecture.
As a result:
• All applications are natively connected and do not require custom integrations to facilitate interoperability.
• A common, harmonized and configurable data model:
• Eliminates data redundancy
• Ensures data accuracy and consistency across all applications
• Custom interfaces to link related records together become immaterial as all information is inherent to all unified applications
Because all applications involved in managing the process share a common UX, users can focus on completing their assigned work and not on mastering the nuances of multiple specialized systems:
• Common activities such as navigation, searching, item creation, and task acquisition are performed identically – regardless of context.
• Cross-organizational communications and collaboration are enhanced.
• Process and information silos are eliminated.
• The “how” questions are answered consistently.
• Total transparency into regulatory processes is achieved – eliminating bottlenecks while ensuring timelines are met and commitments fulfilled.
This unified and ubiquitous approach to human-computer interaction results in higher user adoption rates, lower training requirements, and overall increased productivity.
Finally, the unified platform reduces the overall cost of compliance through the reduction of annual software support and maintenance fees, faster employee ramp-up and lower re-validation costs following system upgrades.
The following figure illustrates the concept of the unified Quality and Regulatory Information Management solution. The users can seamlessly navigate the information hierarchy from the initial change control, through the regulatory activity plan and product information and to the effected documents and related regulatory submission dossiers.

Conclusion
ICH Q12 and the variation change control processes that must be implemented to comply with it is a very clear example of why Life Science companies need not only to move from paper to electronic processes, but also do this without creating information silos between their different information systems.
This would support LCM and ICHQ12 implementation. ICHQ12 is indeed based on the principle to have a robust QMS. Critical information systems for Quality and Regulatory Affairs such as QMS, EDMS and RIM must be either seamlessly integrated, or even better should be part of a unified platform that makes costly and often incomplete integrations obsolete.
Partager l’article


