


Sommaire
- Quelques bonnes pratiques de validation des procédures analytiques
- ALCOA… avec un A pour Accuracy
- Established Conditions for Analytical Procedures and QbD: current situation and future perspectives for enhanced change management paradigm
- Quand les vibrations des molécules permettent de les doser : la spectroscopie proche infrarouge en action
- Apport des études de stabilités accélérées prédictives (APS) au développement pharmaceutique
- Mise en place de la chimie analytique verte au laboratoire contrôle qualité de la société UPSA
- Évaporation des solutions à base d’alcool : Quels résidus sur les équipements ?
Quelques bonnes pratiques de validation des procédures analytiques
Cet article a pour objectif de rappeler quelques fondamentaux de validation des méthodes physico-chimiques mises en œuvre sur des principes actifs ou des produits finis. Il n’aura toutefois pas la prétention de répondre à tous les cas de figure auxquels un laboratoire de R&D ou de contrôle pourra être confronté.
Les notions d’erreur totale seront développées plus en détail dans l’article suivant (“ALCOA… avec un A pour Accuracy”) dans cette revue.

1. Introduction
La validation d’une procédure analytique, qui correspond à l’évaluation approfondie de ses performances (ou de son erreur potentielle), consiste à vérifier, déterminer et estimer plusieurs critères de validation (ou caractéristiques)1 parmi lesquels :
• la spécificité,
• la fonction de réponse (ou courbe d’étalonnage) si applicable,
• la justesse (nommée “accuracy” dans le texte ICH Q2 (R1)),
• la fidélité qui inclus la répétabilité et la fidélité intermédiaire (la reproductibilité étant généralement évaluée lors d’études de qualification de plusieurs laboratoires),
• la linéarité des résultats,
• la limite de détection/de quantification dans le cadre de la quantification des impuretés/ traces/produits de dégradation,
• l’intervalle de travail (appelé aussi “range”).
Cette étape de validation n’est qu’une confirmation que la méthode développée répond aux critères fixés par l’utilisateur pour l’usage qui en sera fait en routine. Elle n’intervient en effet qu’après un développement complet et adapté à ce même usage. En amont de la validation, il aura été nécessaire :
• d’évaluer, comprendre et limiter au maximum les sources de variabilité de la méthode analytique (en particulier au travers d’une ou plusieurs études de robustesse) pour les
méthodes quantitatives
• d’assurer la spécificité de la méthode avec une étude de dégradations forcées
• de prévalider la technique pour s’assurer qu’elle répondra bien aux critères d’acceptation qui seront fixés dans le protocole de validation (et compléter le développement si ce n’était pas le cas)
Il est en effet inutile de valider une méthode non maîtrisée ou pour laquelle le caractère “stability indicating”, s’il est requis, n’est pas assuré. Le développement de la procédure analytique reste une étape cruciale pour une bonne robustesse de la méthode et pour éviter les pertes de temps lors de la validation.
La validation sera alors une confirmation, pour le laboratoire utilisateur et pour les autorités de tutelle, que les performances de la méthode sont en adéquation avec l’usage qui sera fait de la méthode.
Il faut garder en mémoire que l’objectif de toute méthode analytique est de démontrer la Qualité du produit analysé, en particulier en termes d’efficacité (fiabilité du dosage) et de sécurité du patient (détectabilité et quantification des impuretés, substances apparentées et produits de dégradation potentiellement toxiques).
2. Attentes des autorités et besoins scientifiques
Il est intéressant de regarder chaque critère de validation en gardant en mémoire l’impact que cela pourrait avoir sur l’évaluation de l’efficacité du produit ou de la sécurité du patient, tout en veillant de respecter les attentes des autorités : ICH Q2R1[1]. BPF[2, 3], USP[4], guide FDA[5].
Toutefois, il est rarement suffisant de se limiter à “cocher” les cases des requis réglementaires.
Un examen approfondi des résultats de validation est nécessaire pour que l’utilisateur adapte la surveillance de sa méthode selon les performances mesurées en validation.
Ainsi, au lieu de se dire “la méthode est validée, donc mes résultats sont bons», il est vivement conseillé de se donner les garanties que “mes résultats sont fiables, donc ma méthode est valide”.
Cela revient à considérer que la connaissance de la méthode et de ses performances, et la surveillance de ces performances permet d’être confiant en permanence sur le fait que les résultats produits par la méthode sont (et demeurent) fiables. Cette vision du cycle de vie de la méthode, avec la validation comme simple étape parmi tant d’autres, est clairement définie dans le draft USP <1220>[6] et le guide FDA[5].
La fiabilité d’une mesure dépend des erreurs potentielles liées à cette mesure. Cette erreur peut se décomposer en 2 parties distinctes :
• L’erreur systématique (correspondant à la justesse/ “trueness”). Elle se définit comme l’écart de la moyenne de plusieurs valeurs obtenues par rapport à la valeur considérée comme vraie (cette valeur vraie n’est souvent qu’une estimation car elle est rarement connue de manière exacte).
• L’erreur aléatoire (correspondant à la fidélité/ “precision”). Elle se définit comme l’erreur sur chaque valeur individuelle, due aux différentes sources de variabilité de la procédure analytique (et qu’on aura cherché à réduire au maximum durant le développement). Autrement dit, il s’agit de la dispersion des résultats que l’on obtiendra si l’on refait l’analyse x fois.
L’illustration de ces 2 types d’erreurs est fréquemment représentée sous forme de cibles (Figure 1) :
• Au centre : la valeur vraie
• Autour du centre, des limites (critères d’acceptation en validation, seuil d’alerte, limite de spécifications, etc.)
• En rouge, les mesures effectuées
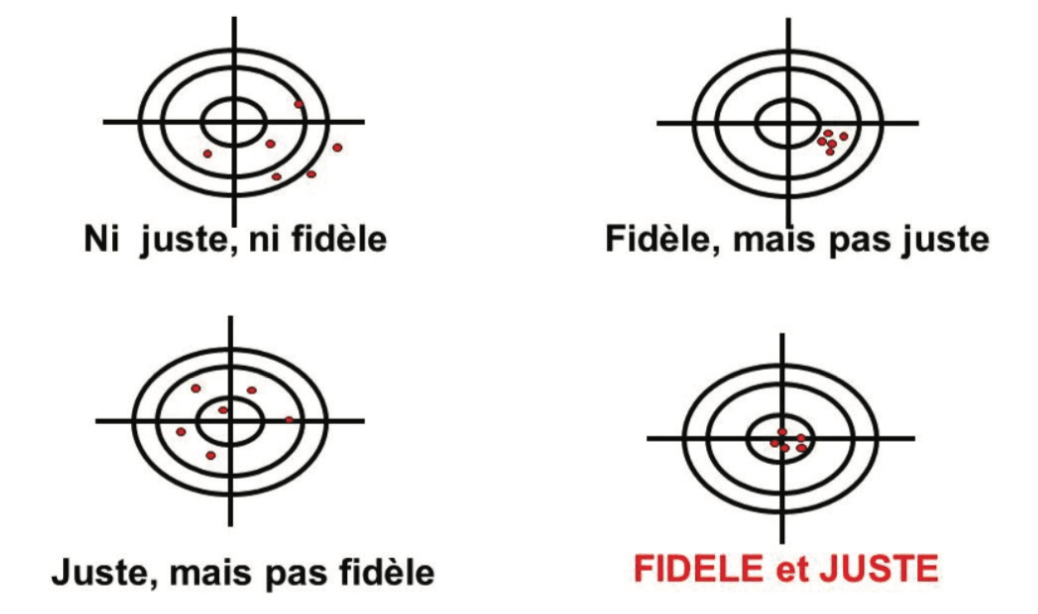
Des résultats fidèles et justes seront entachés d’une incertitude plus faible, et seront donc fiables. Sinon, il faut avoir conscience de l’erreur associée aux mesures obtenues avec la méthode, en lien avec l’incertitude de chacune des mesures. Dans la cible de la figure 1, un point rouge en dehors de la cible illustre le “cauchemar” des laboratoires de contrôle Qualité = résultat OOS lié à la variabilité analytique, pour lequel il est compliqué d’assigner une cause formelle.
En résumé, valider une méthode d’analytique revient à démontrer une capacité à mesurer, quantifier, qualifier, caractériser de façon fiable.
3. Identifier les sources de variabilité et en mesurer l’impact
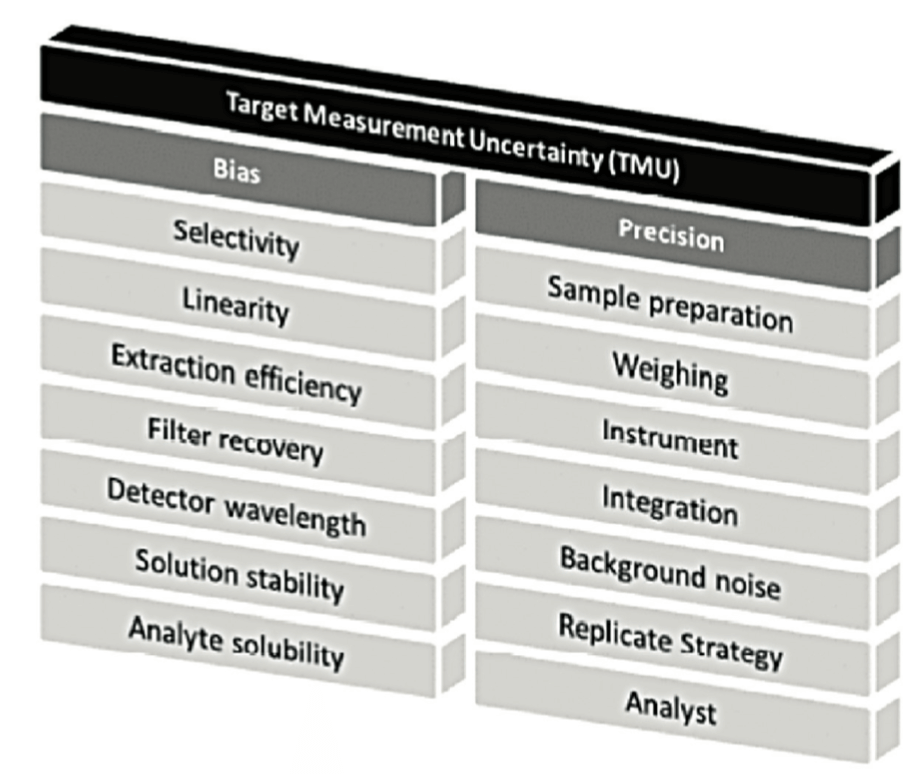
L’incertitude de mesure peut provenir de nombreuses sources (Figure 2 et Figure 3). Il est important d’identifier les sources de variabilité, puis d’optimiser les conditions opératoires pour réduire au maximum leur impact lorsqu’elles sont détectées.
On peut voir, dans cette Figure 2, que différents critères de validation décrits dans ICHQ2(R1) sont étudiés car ils sont de potentielles sources d’erreurs aléatoires ou systématiques. Mais au final, c’est l’impact global sur l’incertitude du résultat qu’il faut considérer. L’article sur le A de ALCOA, dans la présente revue, approfondira ces notions d’erreur totale, si importantes pour appréhender au mieux la fiabilité des mesures effectuées au laboratoire.
4. Design et documentation
Le processus de validation analytique fait suite au développement de la méthode et inclut une partie expérimentale et une partie documentaire qui permettent d’assurer la traçabilité des données. En particulier un protocole et un rapport doivent être écrits et signés respectivement avant et après la validation. Ces 2 documents doivent faire l’objet d’un suivi des différentes versions et des différents changements.
Le protocole doit contenir a minima :
• les critères de validation qui seront évalués (définis en fonction du but de la méthode) et/ou le design choisi,
• la méthodologie pour réaliser ces tests,
• la référence de la méthode à valider et sa version,
• ainsi que les critères ou limites d’acceptation de chaque critère (incluant les attentes en termes de traitements statistiques et/ou graphiques).
On pourra aussi faire référence dans le protocole aux études de développement dont les résultats seront repris dans le rapport de validation (robustesse et dégradations forcées en particulier)
Le rapport doit lui contenir a minima :
• La référence de la procédure validée ainsi que sa version.
• Le cas échéant, les résultats d’études de développement qui complètent la validation.
• Les différentes déviations rencontrées durant la validation.
• Les résultats de validation et les conclusions sur les performances de la méthode.
• Les données brutes pertinentes (résultats, chromatogrammes types).
• Et les éventuelles modifications à effectuer sur la procédure analytique (compléments d’information en particulier).
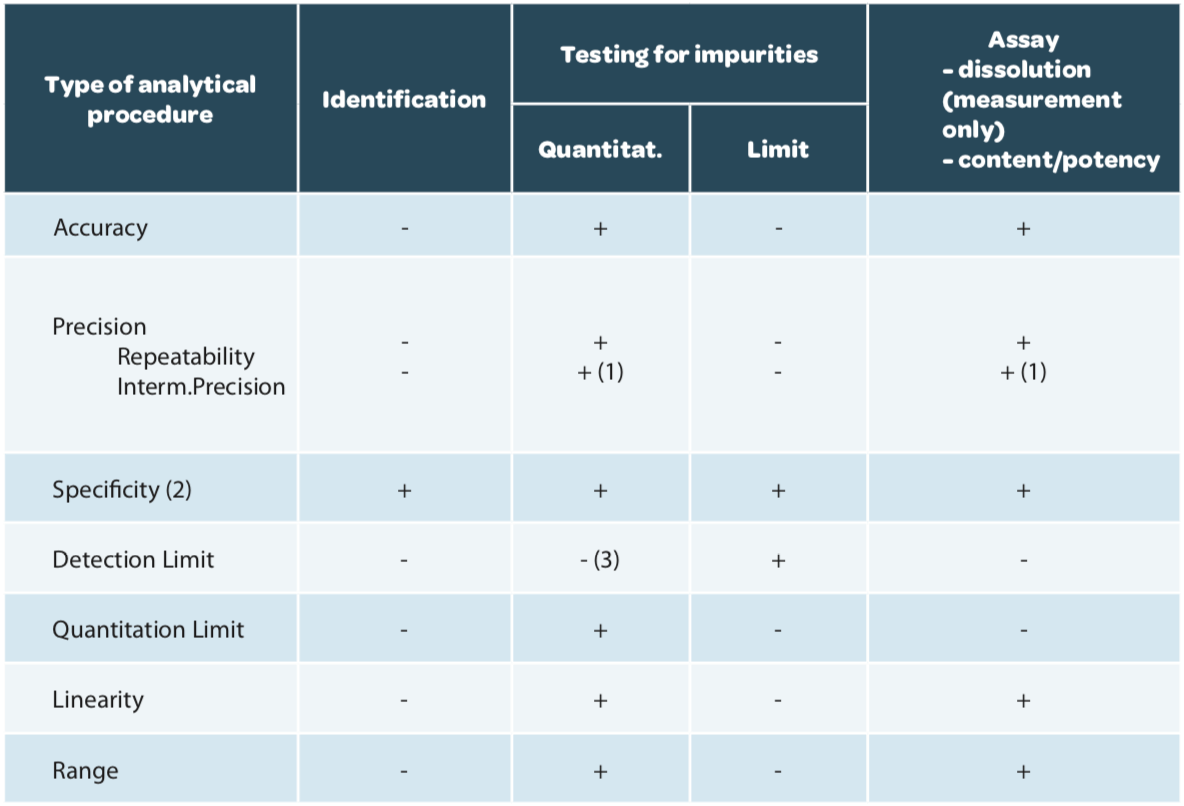
Le design de validation, c’est-à-dire les critères à vérifier, sont décrits dans l’ICH Q2(R1) en fonction du but de la méthode (Tableau 1).

Spécificité / Sélectivité
Le terme “Spécificité” est aujourd’hui utilisé pour les deux termes.
Cependant, on utilise souvent le terme de “sélectivité” pour une méthode qui fournit une réponse pour plusieurs analytes (méthodes séparatives en particulier).
La spécificité d’une procédure analytique est donc sa capacité à évaluer sans équivoque l’analyte, c’est-à-dire à vérifier que rien n’interfère avec la réponse de l’analyte (ou alors de manière non significative).
Pour vérifier la spécificité, on analyse a minima : (i) un blanc, (ii) un placebo (mélange d’excipient sans l’analyte d’intérêt), (iii) un standard et (iv) un produit fini. On pourra également inclure des impuretés spécifiques si nécessaire. On vérifie ensuite que dans le placebo, aucune interférence significative n’est observée.
Le pourquoi et le comment
L’absence d’interférence au niveau de la mesure de l’analyte est évaluée afin de garantir que la quantification ne comportera pas d’erreur. Cette erreur pourra être systématique (interférence stable dans le temps) ou aléatoire (si le composé interférant peut être présent à des concentrations variables).
Il est tout à fait possible de tolérer une interférence si cette dernière est assez faible pour ne pas générer un biais inacceptable (moins de 1 ou 2 % du signal à la cible, par exemple). Mais il convient de s’assurer que l’interférence est constante (qu’elle ne varie pas d’une production à l’autre ou qu’elle n’évolue pas durant une étude de stabilité).
Si une méthode n’est pas spécifique (cas des méthodes de dosage titrimétriques ou des dosages par UV direct), il convient d’avoir une autre méthode qui permette de mieux cerner la composition du produit testé (tests additionnels de pureté par méthode séparative).
Linéarité (des résultats)
La linéarité devrait s’entendre comme la linéarité de la procédure analytique, c’est-à-dire sa capacité, dans l’intervalle de dosage considéré, à fournir des teneurs expérimentales strictement proportionnelles à la teneur en analyte dans l’échantillon.
Il ne faut pas confondre la linéarité avec la fonction de réponse (appelée aussi courbe d’étalonnage) qui représente la réponse analytique en fonction de la concentration du/des standard(s). Or, il est tout à fait possible que la fonction de réponse ne soit pas linéaire (par exemple, avec un détecteur ELSD, pour un dosage Elisa..) mais que la linéarité des résultats soit bien assurée. C’est bien cela qui est recherché.
Pour vérifier la linéarité, il convient de tester plusieurs niveaux de concentrations répartis dans l’intervalle de dosage. On trace ensuite le graphe de la concentration de l’échantillon déterminé expérimentalement en fonction de la concentration théorique.
Les critères sur les résultats de la régression linéaire peuvent par exemple être d’avoir une droite de pente proche de 1 et une ordonnée à l’origine proche de 0 avec un coefficient de détermination supérieur à 0.99.
Or, le guide ICH Q2(R1) n’exige qu’un critère sur le coefficient de corrélation, un graphe et l’équation de la régression, ainsi que la somme des carrés des résidus, calculés lors de la régression. Nous vous rassurons, ce dernier paramètre est rarement exigé dans les rapports car son interprétation n’apporte pas de valeur ajoutée à l’objectif d’une validation de procédure analytique.
Le pourquoi et le comment
L’intérêt de ce paramètre est d’identifier un éventuel effet de la matrice, si l’étude est faite sur un reconstitué. C’est pour cela que l’intérêt d’effectuer des linéarités en solution est fort limité, sans cette étude sur reconstitués.
Seule cette linéarité des résultats est importante pour garantir la fiabilité des analyses et est totalement liée à l’étude de la justesse, comme l’indique la figure 2.
Pour l’étalonnage, si les réponses obtenues avec la méthode ne sont pas linéaires, il est alors possible de déterminer une autre fonction mathématique (pondérations, modèle quadratique, log/log ou autres transformations) pour obtenir des résultats linéaires.
Justesse
La justesse exprime l’étroitesse de l’accord entre la valeur moyenne obtenue à partir d’une série de résultats d’essais et une valeur qui est acceptée soit comme une valeur conventionnellement vraie, soit comme une valeur de référence acceptée (ex : standard international, standard d’une pharmacopée, résultat obtenu par une autre méthode validée). Il s’agit d’une mesure de l’erreur systématique (biais).
Pour la détermination de la justesse, conformément à ICH Q2(R1) dans le paragraphe “Accuracy”, il est recommandé de le faire a minima avec 3 niveaux de concentration et 3 réplicas par niveau (9 déterminations). Ces résultats serviront également pour la détermination de la linéarité. Pour l’évaluation de la justesse (et de linéarité), il convient de créer des échantillons reconstitués dont la concentration de l’analyte est connue dans la même matrice que le produit qui sera analysé. Il est parfois délicat d’accéder à ces reconstitués qui soient préparés avec un processus similaire à celui utilisé pour le produit fini. Les reconstitués produits au laboratoire peuvent parfois engendrer des problèmes d’homogénéité qui conduisent à des conclusions erronées sur les performances de la méthode.
Dans le cadre de comprimés ou autres formes galéniques solides ou semi-solides, une solution consiste à ajouter une quantité connue d’analyte sur un mélange d’excipients. Cette voie ne permet pas de mimer parfaitement le mécanisme d’extraction de l’analyte de la matrice. Ce phénomène d’extraction pourra cependant être évalué
partiellement lors des études de fidélité même grossièrement (en vérifiant que la teneur obtenue pour chaque préparation est bien celle attendue).
La justesse est exprimée en termes de pourcentage de recouvrements, c’est-à-dire que l’on calcule pour chaque niveau le recouvrement moyen (quantité calculée/quantité ajoutée, en tenant compte éventuellement de la quantité initiale si on n’a pas de composé sans l’analyte recherché, comme dans le cas des solvants résiduels). En effet, pour chaque détermination on calcule le ratio entre la teneur déterminée expérimentalement par la teneur déterminée théoriquement. On effectue ensuite la moyenne des recouvrements. Les recouvrements de chaque niveau doivent être inclus dans les critères d’acceptation pour la justesse. Limiter le raisonnement sur la moyenne de tous les niveaux pourrait en effet masquer des disparités entre niveaux (il conviendrait alors de limiter la dispersion des données).
Il convient également de fournir les intervalles de confiance de la moyenne, conformément à ICH Q2(R1)
Le pourquoi et le comment
La vérification de la justesse est importante. Elle permet de vérifier qu’à chaque niveau de concentration, la procédure analytique va mesurer avec justesse la teneur en analyte (ou, pour reprendre l’image des cibles, si on bouge la cible, on continuera à tirer dedans et pas à l’endroit où était la cible auparavant). La moyenne des recouvrements sur les réplicas de chaque niveau permet de détecter un biais sur un des niveaux. Il est à noter que l’analyse de la linéarité de la méthode et des résultats doit mener à la même conclusion que l’étude de la justesse.
En fonction des niveaux de concentrations, il sera possible de proposer des cibles plus ou moins larges. En effet, plus la concentration est faible, plus l’incertitude de mesure augmente, et il est communément admis d’élargir les critères d’acceptation au point bas du range étudié.
Fidélité
La Fidélité (“precision” en anglais) d’une procédure analytique exprime l’étroitesse de l’accord (mesure de la dispersion) entre une série de mesures obtenue à partir de plusieurs prises d’essai d’un même échantillon homogène, dans les conditions de la procédure analytique.
C’est une mesure de l’erreur aléatoire qui s’évalue selon plusieurs conditions :
- La répétabilité.
- La Fidélité intermédiaire.
- La reproductibilité.
Ces conditions sont schématisées par des couches de “l’oignon de la variabilité” présenté dans la Figure 3.
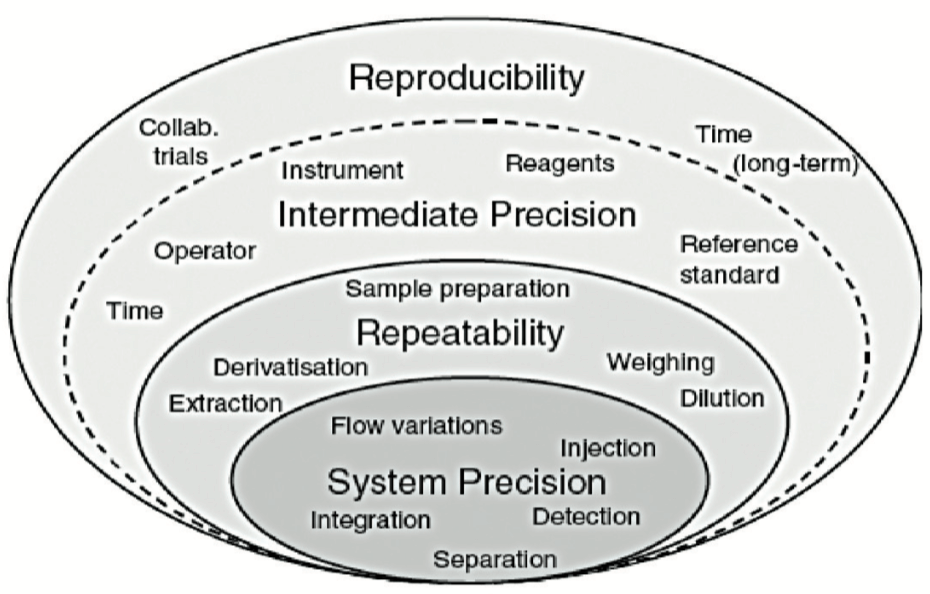
Chaque couche dans cette Figure 3 se caractérise par des sources de variabilité différentes :
• La première couche correspond aux sources de variabilité du système utilisé. La qualification des équipements doit assurer une couche aussi faible que possible à ce niveau-là.
• La deuxième couche “répétabilité” inclut essentiellement la variabilité de la préparation d’échantillon.
• La troisième couche “Fidélité intermédiaire” inclut les sources de variabilité entre différents étalonnages, différents jours, instruments, différents techniciens, mais potentiellement aussi des variations saisonnières, rarement évaluées en validation (température du laboratoire entre été et hiver, quantité de lumière passant par les fenêtres du laboratoire, etc..).
Une 4ème couche, généralement non évaluée en validation, inclut la variabilité entre laboratoire. Elle est généralement évaluée (ou subie, si on n’y prend pas garde) lors des transferts analytiques ou lors de co- validation entre 2 laboratoires.
La mesure de la Fidélité est souvent effectuée par la détermination des coefficients de variation pour la répétabilité et la Fidélité intermédiaire. La répétabilité est évaluée (selon ICH Q2(R1) avec :
• 9 déterminations répartis sur 3 niveaux et 3 réplicas par niveau (a minima)
ou
• 6 déterminations à 100% de la cible de validation.
Pour évaluer la Fidélité intermédiaire, il faut multiplier les séries en prenant soin de modifier le technicien et/ou le système utilisé et/ou le jour de la mesure. Un minimum de 3 séries est nécessaire pour pouvoir estimer la variabilité entre les séries.
Les coefficients de variation de la répétabilité et de la Fidélité intermédiaire sont déterminés via une analyse de type ANOVA (analyse de variance).
Pour faire ces calculs il est nécessaire de déterminer les variances de répétabilité et de Fidélité intermédiaire (incluant des variabilités INTRA série et INTER série) puis d’en déduire les coefficients de variations. Les définitions sont données dans le Tableau 2.
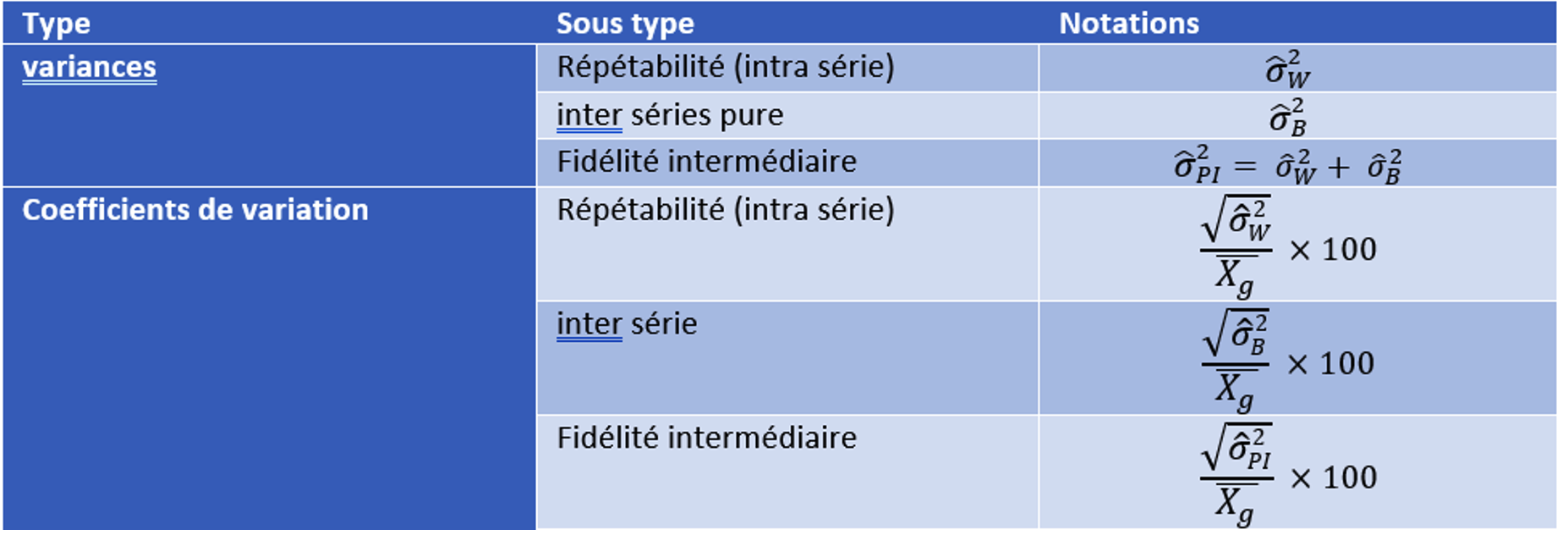
La méthode pour la détermination des coefficients de variation de répétabilité et de Fidélité intermédiaire se fait selon la méthodologie présentée dans le Tableau 3 pour p séries à n valeurs par série.
 Tableau 3 : calcul du coefficient de variation pour la répétabilité et la fidélité intermédiaire par une analyse de type ANOVA. On considère p séries à n valeurs par série et Xj les moyennes des séries j et Xg la moyenne globale de toutes les valeurs.
Tableau 3 : calcul du coefficient de variation pour la répétabilité et la fidélité intermédiaire par une analyse de type ANOVA. On considère p séries à n valeurs par série et Xj les moyennes des séries j et Xg la moyenne globale de toutes les valeurs.
Attention, les formules de calcul présentées dans le Tableau 3 ne sont valables que que s’il y a le même nombre de répétition par série et dans le cas d’un design avec des préparations indépendantes de mêmes cibles.
Le pourquoi et le comment
L’objectif est d’estimer la variabilité de la méthode qui sera rencontrée en routine, durant le cycle de vie de la méthode. Il n’est donc pas judicieux de travailler “exceptionnellement bien” durant la validation, avec le risque de sous-estimer cette variabilité.
Cela reviendrait à minimiser le risque analytique réel, comme souligné dans l’article “ALCOA… avec un A pour Accuracy” dans ce même journal.
Trop souvent le CV de répétabilité est calculé uniquement sur la première série. Dans la méthode proposée, le CV est représentatif de toutes les valeurs générées. En effet, il est important d’estimer le CV de répétabilité sur l’ensemble des valeurs (via les variances de chacune des séries) pour avoir une estimation au plus proche de la vérité. Ce CV est représentatif des 2 premières couches de l’oignon de la variabilité présenté dans la Figure 3.
De même, le CV de fidélité intermédiaire est parfois calculé comme l’écart-type de toutes les valeurs par rapport à la moyenne générale (sans tenir compte du fait qu’elles proviennent de séries différentes). Ce calcul est erroné et ne rend pas compte de la variabilité inter séries, ce qui a pour conséquence de minimiser le coefficient de variation de la fidélité intermédiaire. Ce CV est représentatif des 3 premières couches de l’oignon de la variabilité présenté dans la Figure 3.
Il est donc impossible que le CV de fidélité intermédiaire soit inférieur au CV de répétabilité. Au mieux, l’épaisseur de la 3eme couche est infime, mais elle ne peut en aucun cas diminuer l’épaisseur de la 2ème couche.
Limite de détection (LOD)/ Limite de quantification (LOQ)
L’analyse des limites d’une méthode est essentielle pour se donner les garanties qu’on est bien capable d’analyser l’analyte recherché en deçà de la limite acceptable pour cette impureté. Pour les méthodes quantitatives, il faudra garantir qu’on peut quantifier de façon fiables tous les résultats rapportables (au-dessus du “reporting threshold”, défini selon la posologie, comme décrit dans la série des guides ICHQ3 [9, 10, 11, 12]).
La limite de détection se définit comme la plus petite teneur de l’analyte provenant d’un échantillon pouvant être détectée. La limite de quantification est la plus petite teneur de l’analyte pouvant être dosée de manière exacte (combinaison des erreurs systématique et aléatoire).
La détermination de la LOD n’est requise que pour les essais limites alors que la LOQ est déterminée pour les dosages d’impuretés ou de traces. Quelle que soit la méthode de détermination utilisée, il conviendra de vérifier la fiabilité des mesures à cette limite [5].
La détermination des LOD et LOQ peut se faire selon plusieurs méthodes décrites dans le guide ICH Q2(R1). Chacun peut choisir son mode de calcul. Ces différentes méthodes ne donnent pas toujours le même résultat, mais l’essentiel reste de garantir que la LOQ sera inférieure ou égale au “reporting threshold”.
Le pourquoi et le comment
Il n’est pas forcément nécessaire de rechercher la limite la plus basse possible. En effet, l’”Intended Use” est de rapporter des valeurs à partir du “Reporting Threshold”. Valider à des niveaux inférieurs n’apporte donc pas de valeur ajoutée. Il est donc possible de définir la “Working LOQ” comme étant le point bas de notre intervalle de dosage souvent définis par les limites de report décrites dans les guides ICH Q3. Ainsi, ce niveau bas de concentration sera bien validé en termes de justesse et de fidélité, comme attendu par les autorités.
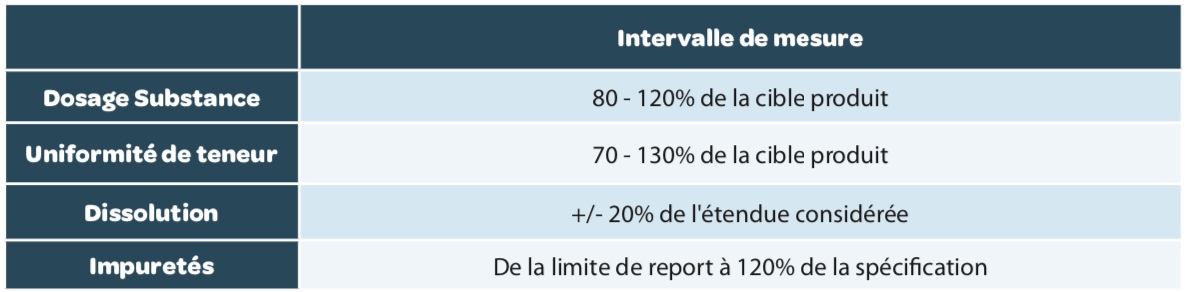
Intervalle de mesure
L’intervalle de mesure est l’intervalle dans lequel la méthode assure un degré acceptable de linéarité, justesse et fidélité pour toutes les mesures dont les résultats sont compris dans cet intervalle. Généralement, les intervalles de mesure attendus sont résumés dans le Tableau 4 mais peuvent être plus large si cela est nécessaire.
Combinaison de l’erreur systématique et de l’erreur aléatoire
L’exactitude (au sens ISO du terme) est le paramètre qui inclut à la fois l’erreur systématique et l’erreur aléatoire (justesse + fidélité), bien que les notions de justesse et d’exactitude soit parfois confondues dans certains textes réglementaires. Il est donc plus adapté, dans le domaine pharmaceutique, de parler d’erreur totale (ou d’erreur combinée).
Il s’agit de la vraie finalité d’une validation de procédure analytique, qui est de garantir la fiabilité de chacune des mesures (au sens individuel) qui seront obtenues ultérieurement. L’incertitude de mesure et la notion d’erreur totale sont intimement liées.
Un autre intérêt de cette démarche est qu’un potentiel problème d’erreur systématique important peut être compensé par une erreur aléatoire faible et vice-versa.
Ces notions d’erreur totale sont développées dans l’article suivant de cette revue (“ALCOA… avec un A pour Accuracy“).
Stabilité des solutions
La stabilité des solutions, souvent incluse dans l’étude de robustesse, est importante pour garantir que les données générées restent exactes et permet de connaitre les temps de conservation de ces dernières.
La stabilité doit être effectuée sur les solutions :
• De standards (solutions mère et fille dans les fioles et dans les flacons).
• D’échantillons.
• De phases mobiles.
La stabilité des solutions s’entend au regard du but de la procédure analytique.
Pour un dosage, il s’agira de vérifier que la teneur déterminée ne varie pas au-delà de la variabilité analytique classique. Pour la recherche d’impuretés, il faudra regarder attentivement les variations des profils chromatographiques, et vérifier qu’aucune nouvelle impureté n’apparait ou qu’aucune impureté présente dans l’échantillon ne disparait au fur et à mesure des analyses.
L’étude consiste à effectuer une mesure de chaque solution considérée (certains laboratoires faisant des répétitions de ces solutions) à différentes échéances, et de la comparer par rapport à un nouveau standard de référence. L’écart par rapport à la valeur initiale est alors calculé et doit être conforme aux critères d’acceptation définis. Ces critères sont à définir en fonction de ceux définis pour la justesse et la fidélité
On peut citer une autre pratique, pour les méthodes séparatives, qui consiste à suivre l’aire des pics d’une même solution à intervalle régulier. L’absence d’évolution significative des aires est une autre façon de garantir que la fiabilité des mesures n’évolue pas avec l’âge des solutions analysées. Cela ne sera possible que si l’étude est faite en continu, sans arrêter le système d’acquisition, afin de comparer des valeurs comparables.
Critères/limites d’acceptation
Les critères/limites d’acceptation de la validation de la procédure analytique sont à définir en fonction de plusieurs critères et notamment :
• Le domaine d’application (pharmaceutique, agroalimentaire, cosmétique, “medical device”…).
• Les spécifications du produit, définies dans le dossier. Ces spécifications incluent une marge pour la variabilité de production, des prélèvements et la variabilité analytique. Il convient donc de bien partager l’espace “disponible” entre les limites de spécifications entre la production et l’analytique et de se garder une marge de sécurité pour éviter les potentiels résultats OOS du fait de la variabilité analytique.
• L’usage qui sera fait de la méthode et des résultats qui en seront issus.
Beaucoup de critères descriptifs empiriques existent dans le domaine pharmaceutique. Nous pouvons conseiller le visionnage de la vidéo YouTube partagée par Oona McPolin qui explique l’origine de certains des critères classiquement rencontrés.
Nous ne fournirons pas d’exemples dans cet article, car ils ne pourraient être que des exemples, qui doivent être adaptés au contexte de votre analyse, et aux performances déjà connues de votre méthode.
On peut toutefois mettre l’accent sur certaines erreurs classiques à éviter :
• Critères statistiques inadaptés. Certains tests statistiques (Cochran, Fisher, Student) peuvent vous apporter des informations sur les données générées en validation. Toutefois, du fait du faible nombre de données générées, ces tests ne sont pas toujours concluants. Et ils sont rarement la preuve que votre méthode génère des résultats fiables ou pas. Utiliser ces tests était une pratique courante, mais il est vivement conseillé de ne pas utiliser les conclusions de ces tests comme étant des critères d’acceptation.
• Critères trop larges. Si cela pouvait “faciliter” la rédaction d’un rapport de validation, avoir des critères trop larges ne permettra pas de garantir la fiabilité des mesures qui seront générées ensuite.
• Critères trop étroits (à mauvais escient). Un exemple de “mauvaise” pratique parfois constatée est un critère de biais systématique sur des impuretés fixé très bas dans le protocole de validation, sans tenir compte des biais qui seront apportés ensuite en arrondissant les résultats (par exemple : biais maximum = 5% pour une impureté devant être inférieure à 0,5%, alors que le fait d’arrondir un résultat obtenu à 0.25% à 0.3% représente à lui seul un biais de 20%). Un autre exemple concerne le niveau d’interférence acceptable lors de l’étude de spécificité. Comme signalé dans la Figure 2, une interférence peut engendrer un biais systématique. Mais si, par exemple, un biais systématique total de 2% est accepté, il ne parait pas logique de limiter alors chaque interférence à un niveau extrêmement faible (0.05%, par exemple). Mais si un critère est fixé de façon étroite pour des raisons de sécurité de patient (et donc à bon escient), nous ne pouvons que vous encourager à faire ainsi.
Des limites devront donc être fixées pour chacun des critères de validation, mais sans oublier qu’ils sont inter-reliés, comme illustré en Figure 2. Les limites fondamentales sont donc celles fixées sur le l’erreur systématique et sur le l’erreur aléatoire, les 2 piliers de l’incertitudes. Les autres limites permettent de mieux cerner certaines sources de ces 2 erreurs à caractériser.
La combinaison des 2 erreurs par l’erreur totale (combinée) reste le meilleur moyen de contrôler la fiabilité des mesures qui seront générés. Parmi les critères sur la combinaison des 2 sources d’erreur, on peut retrouver des critères sur la capabilité d’une méthode ou des graphes incluant des courbes de niveaux de risques (Operating characteristic curves) [12, 13], qui sont des moyens simples pour estimer le niveau de risque analytique, sans toutefois permettre une réelle gestion de ces risques.
La tendance actuelle va vers le profil d’exactitude et son intervalle de Tolérance [13] ou de l’intervalle de Prédiction [4] qui restent des outils statistiques un peu plus complexes mais plus puissants car ils permettent de gérer et contrôler le risque analytique.
Dans tous les cas, la détermination de ces limites d’acceptation est cruciale et doit être effectuée, certes pour répondre aux exigences réglementaires, mais également pour s’assurer que la procédure analytique sera scientifiquement performante, c’est-à-dire qu’elle sera juste et fidèle (voir Figure 1).
5. Après la validation
Après qu’une méthode a été validée, il est vivement conseillé de la mettre “sous surveillance”.
La surveillance au quotidien réside dans la conformité des critères fixés dans les tests de conformité du système d’analyse (SST= System Suitability test). Selon les techniques et l’objectif des méthodes, ces tests incluent des critères de qualité du signal (symétrie de pics, etc…) des critères de bon fonctionnement de l’équipement (répétabilité des mesures en particulier), des solutions de contrôle, des critères chromatographiques pour les méthodes séparatives, etc.
Outre la carte de contrôle classiquement mise en œuvre pour les résultats de différents lots produits, la tendance va vers la mise ne place de carte de contrôle des différents paramètres du SST (critère de résolution, symétrie de pic, répétabilité de mesure, suivi de contrôle qualité (interne ou externe), etc) afin d’anticiper sur des dérives ou des dysfonctionnements potentiels, qui seront plus facilement détectables.
Par ailleurs, en cas de changement, il faudra évaluer les besoins de revalidation partielle ou totale selon les besoins. Parmi les changements, non exhaustifs, qui conduisent à une revalidation, l’ICH Q2(R1) [1] cite :
• Les changements de synthèse de la “drug substance” ce qui inclut généralement la source de cette dernière.
• Les changements de composition des produits finis.
• Les changements de procédure analytique.
Un changement de laboratoire est généralement géré sous forme de transfert analytique, qui peut faire l’objet d’études inter-laboratoires ou de revalidation (totale ou partielle) au sein du laboratoire receveur. Dans tous les cas, une connaissance approfondie de la méthode, de ses sources de variabilité à maîtriser et de ses performances sont autant d’atouts pour alimenter vos analyses de risques et limiter ces besoins de revalidation aux seuls tests pertinents au vu du changement prévu.
Conclusion
Comme signalé en préambule, il n’est pas possible de répondre à tous les cas de figure qui peuvent se rencontrer en validation à travers seulement quelques pages.
ICHQ2(R1) insiste sur la responsabilité des détenteurs d’AMM de démontrer, avec un protocole adapté, que la méthode présente des performances suffisantes pour l’usage qui sera fait de la méthode.
Les tendances actuelles[5,6] remettent l’exercice ponctuel de validation à sa place, entre un développement analytique qui permette d’atteindre un niveau de maîtrise adéquat, et une utilisation contrôlée. Nous sommes aussi totalement conscients que certaines exigences régionales iront à l’encontre d’éléments proposés dans cet article. Il convient à chacun d’ajouter à leur protocole de validation les critères ou les tests statistiques spécifiquement requis dans tel ou tel pays.
Le message principal restera qu’au final, la connaissance de l’incertitude du résultat est l’objectif principal d’une validation. Cette incertitude est liée à l’erreur totale, combinaison d’une erreur systématique et d’une erreur aléatoire qui restent indissociables lors des analyses en routine.
L’article qui suit démontrera l’importance de cette erreur totale.
Partager l’article


Eric Chapuzet – Qualilab
Eric est Directeur Général de QUALILAB, société spécialisée dans le conseil en management de la qualité, sur le cycle de vie de méthodes d’analyse, les systèmes d’informations, l’intégrité des données et l’analyse de données. De formation scientifique (statistique, informatique, qualité, physiologie, pharmacologie, biochimie), il participe régulièrement à des projets de validation et transfert de méthodes analytiques. Éric anime et réalise également des formations et des audits dans le cadre des étapes du cycle de vie des méthodes d’analyse depuis de nombreuses années.

Gerald de Fontenay – Cebiphar
Depuis plus de 25 ans, Gérald a assuré différents postes au sein de sociétés de services analytiques et de CDMO. Aujourd’hui Directeur Scientifique et Technique chez Cebiphar, il continue à œuvrer pour garantir le maximum de fiabilité aux résultats qui sont générés dans ses laboratoires. Il apporte son expertise en validation, vérification et transfert de méthodes analytiques auprès des chefs de projets et des clients de Cebiphar, pour assurer le succès des projets qui lui sont confiés.
gdefontenay@cebiphar.com
Marc François Heude – Siegfried
Titulaire d’un doctorat en chimie organique, Marc assure, depuis 6 ans, des rôles de chef de projets analytiques d’abord à Adocia (Société de biotechnologie Lyonnaise) puis à Bayer Healthcare. Il exerce aujourd’hui en tant que responsable du service de développement analytique chez Siegfried, un CDMO de l’industrie pharmaceutique qui produit des APIs et intermédiaires de synthèse.
Références
[1] : ICH Q2 (R1) “Validation of Analytical Procedures: Text and Methodology”, 2005. [2] : FDA: 21CFR : Current Good Manufacturing Practice (CGMP) Regulations, 2020.
[3] : EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines. [4] : US Pharmacopoeia: USP <1210>; <1225>; <1226>.
[5] : FDA Guidance: “Analytical Procedures and Methods Validation for Drugs and Biologics”, 2015.
[6] : Pharmacopeial Forum (PF), 1220 Analytical Procedure Life Cycle — PF 46(5), 2020.
[7] : USP stimuli to revision process 42(5) ANALYTICAL CONTROL STRATEGY, 2016.
[8] : J. Ermer, C. Burgess, G. Kleinschmidt, J. H. McB. Miller, D. Rudd, M. Bloch, H. Wätzig, M. Broughton et R. A. Cox, Method Validation in Pharmaceutical Analysis: A Guide to Best Practice, Wiley, 2005.
[9] : ICH Q3A(R2) IMPURITIES IN NEW DRUG SUBSTANCES, 2006.
[10] :ICH Q3B(R2) IMPURITIES IN NEW DRUG PRODUCTS, 2006.
[11] : ICH Q3C(R8) IMPURITIES:GUIDELINE FOR RESIDUAL SOLVENTS, 2021.
[12] : ICH Q3D(R1) GUIDELINE FOR ELEMENTAL IMPURITIES, 2019.
[13] : P. Hubert, J.-J. Nguyen-Huu, B. Boulanger, E. Chapuzet , P. Chiap, N. Cohen, P. Compagnon, W. Dewé, M. Feinberg et
M. Lallier, «Validation des procédures analytiques quantitatives harmonisation des démarches,» S.T.P. Pharma Pratiques, vol. 13, p. 101, 2003.
[14] : USP <1080> “BULK PHARMACEUTICAL EXCIPIENTS— CERTIFICATE OF ANALYSIS”, 2021.
[15] : Small Molecules Collaborative group and USPC Staff. Pharm Forum 35(3) page 765 – 771, 2009.
[16] : G. de Fontenay, J. Respaud, P. Puig,
C. Lemaire, «Analysis of the performance of analytical method, Risk Analysis for routine use,» S.T.P. Pharma Pratiques, vol. 21, n°12, pp. 123-132, 2011.
