


Summary
- Some good validation practices for analytical procedures
- ALCOA …with an A for Accuracy
- Established Conditions for Analytical Procedures and QbD: current situation and future perspectives for enhanced change management paradigm
- When the vibrations of molecules make their assay possible: near infrared spectroscopy in action
- Input from accelerated predictive stability (APS) studies to pharmaceutical development
- Implementation of green analytical chemistry in the quality control laboratory of the company UPSA
- Evaporation of alcoholic solutions. What residues are left on equipment?
ALCOA …with an A for Accuracy
Generating analytical data is very easy. All it takes is to execute an analysis, obtain an answer, and interpret it by generating a numerical result. Generating a reliable result that is representative of the product we wish to characterise, however, and which provides guarantees regarding its Quality, is a very much more complex process. GMP,[1] like any other quality or regulatory management system, requires qualified equipment, used by trained staff, in a controlled environment. The fundamentals…

With this equipment, a duly validated analytical method must also be implemented. And if this equipment includes a computerized system, IT validation will be required. There is a widespread preoccupation with data integrity, particularly amongst those who are responsible for Quality Assurance. It is indeed important to ensure that data generated by a system are accurate, complete, and not corrupted in any way during their life cycle.[2]
Beyond qualification of the equipment and validation of the systems that generate analytical data, consideration must be given to the analytical method itself, which is the initial source of these data. This analytical method has been developed to characterize one or more Critical Quality Attributes of the product analysed, in order to at least partially guarantee its intrinsic Quality.
During the life cycle of the method, which runs parallel to the life cycle of the product, the reliability of measurement should therefore be known and controlled over the long term. And this measurement reliability is characterized during validation of an analytical procedure through separate analysis of systematic error (bias), and random error (spread/precision), which can be combined via a total error evaluation.[3, 4] The results of this validation study, possibly supplemented by control charts of the performance of the analytical procedure, are therefore the main guarantors of the accuracy of the data generated, in addition to the requirements to create record files.
One of the 2 As in ALCOA (A for “ACCURACY”) is therefore dependent in the first instance on the reliability of the measurements generated, and should highlight the importance of the validity of data, guaranteed by method validation and by everyday safeguards (SST: System Suitability Test) to avoid any deviation by equipment or by the analyst during execution of the analytical procedure.
1. What is data integrity?
In its “GxP Data Integrity Guidance and Definitions” guide published in 2018,[2] the MHRA (Medicines and Healthcare Products and Regulatory Agency) defines integrity as being the assurance that data produced by a system are accurate, complete and have not been corrupted in any way during their LIFE CYCLE.
To ensure data integrity, it should be checked that the data are:
• Accurate – no error or correction without documented modifications.
• Legible – easily read.
• Contemporaneous – documented at the time of the activity.
• Original – all documents or observations are written and printed (or certified copy of the said document).
• Attributable – knowing who generates the data item or carries out an action on it and when.
• Complete – all data are present and available.
• Consistent – all elements in the record, such as chronology are tracked and dated or time stamped in the expected order.
• Enduring – stored on approved storage media (paper or electronic).
• Available – for review, audit or inspection during the lifetime of the record.
The first 5 terms constitute the acronym: ALCOA
The last 4 constitute the + in the acronym: ALCOA +
2. The analytical process
Analysis is a process like any other. Full control of this process will ultimately have as much importance as control of the manufacturing process. A compliant certificate of analysis is indeed just the combination of compliant manufacture and an analysis that is representative of the product tested.
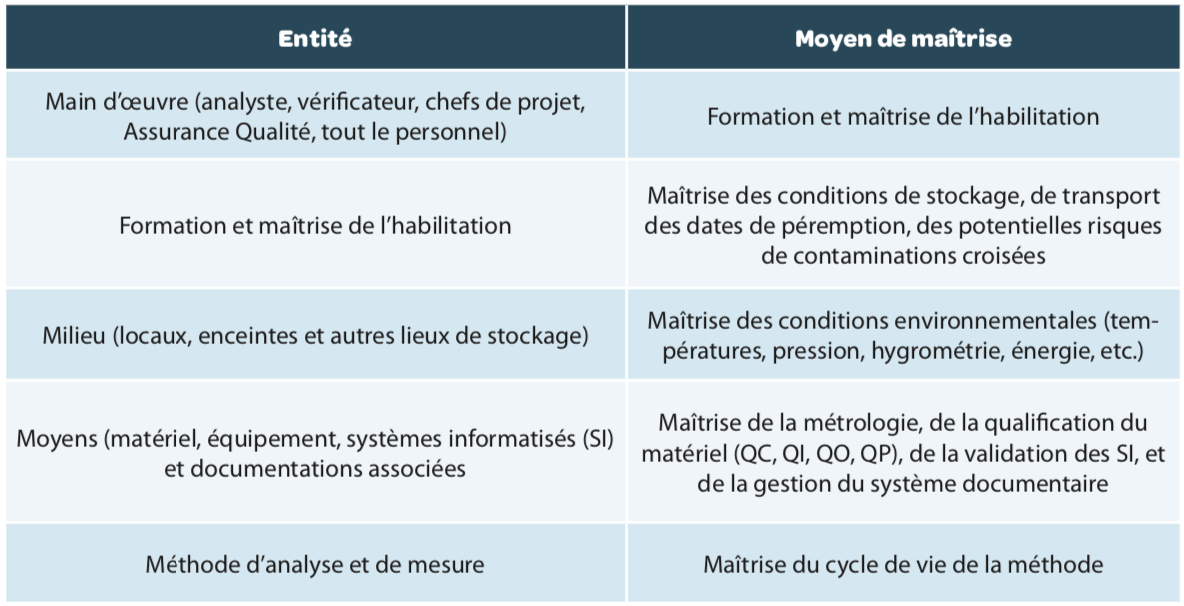
Each entity in this process (Manpower, Materials, Environment, Resources and Methods) must be controlled (Figure 1 and Table 1), that is to say must be compliant with the requirements laid down on its establishment, but also over time.
2. Life cycle of the method
All too often, control of a method is limited to knowing its valid or non-valid status. The recent publication in Pharmacopeial Forum of the draft of USP <1220>[5] will have opened eyes to the importance of all the other steps in this life cycle, already highlighted in the FDA Guidance on validation in 2015. [6]
- Complete analytical development, providing knowledge of the strengths and weaknesses of the method used, allowing all sources of potential variation to be reduced as much as possible (as described in the previous article on “Some Good Validation Practices” in this journal) and defining their conditions of application through a robustness study conducted before validation.
- Validation of the method under the predefined conditions during development, with an in-depthanalysis of the real reliability of the measurements generated by the analytical procedure.
- Monitoring of the analytical procedure, with SSTs (System Suitability Test) specifically defined for the method employed, allowing any error by the analyst or any deviation of the method or the instrument used to generate these data to be avoided or identified.
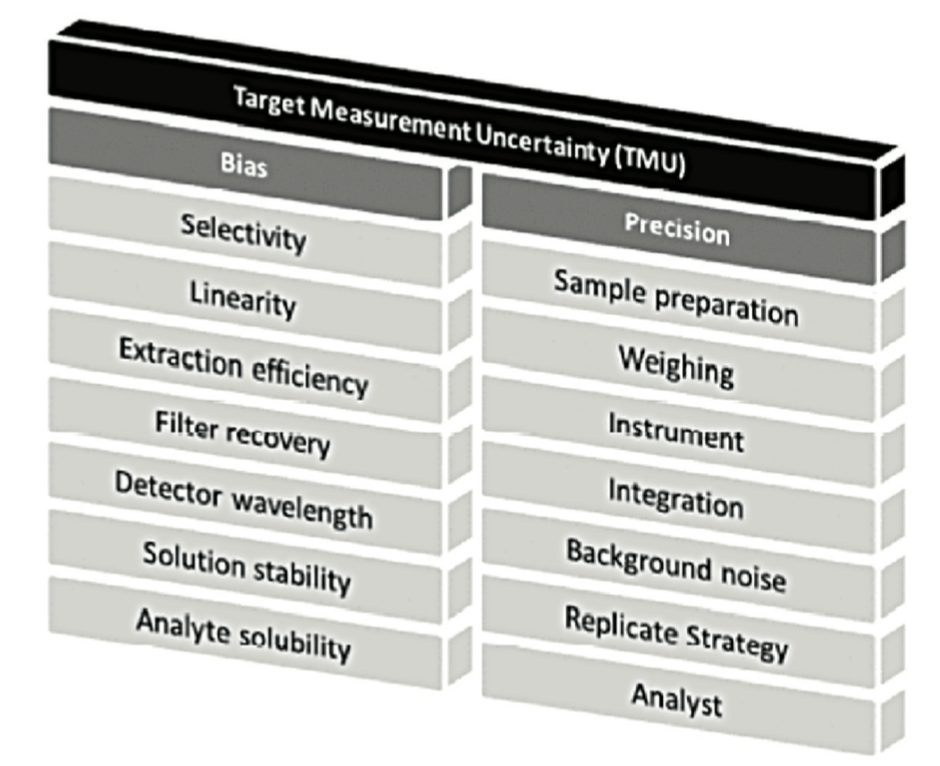
At the heart of this life cycle, method validation remains an important step, however. In a previous version of chapter <1220>, the concept of TMU = Target Measurement Uncertainty was represented by a “temple” with 2 main pillars: “Bias” and “Precision”, as illustrated in Figure 2.
The “Target Measurement Uncertainty” (maximum target uncertainty) can be set as an objective prior to method development.
The developers will therefore ensure that the sum of the different sources of error (systematic error (“Bias”) and random error (“Precision”)) will not impact measurement reliability too much. For this, certain factors will be strictly fixed to minimise their potential impact.
In validation, the main objective will be to measure the residual error that cannot be avoided.
Residual systematic error will be estimated during the study named Accuracy in ICH Q2(R1) [7] (the appropriate term being “Trueness”). Residual random error will be estimated during the study of precision (repeatability, intermediate precision). And the combination of these two sources of error, in the form of an analysis of Total Error or combined error[3, 4] will provide a real grasp of the uncertaintyof each individual measurement,as described in the following section of this article.
A final step will be to define the result that will be reported in the certificates of analysis. This reportable result may be an individual measurement or the mean of several measurements obtained in accordance with a justified and documented strategy, as already described in La Vague No. 68 (article “Replicate strategy”).[8]
4. The “why” of validation
Why do we validate our analytical procedures?
Although many of the questions below may seem simplistic or repetitive, it is still important to keep the answers in mind. And especially not to stop after the first “why”…
Why No. 1. Regulatory requirements
The use of validated analysis methods is a requirement of the health authorities. All the referencetexts (GMP[1], USP [9], ICH Q2R1 and soon Q14 or another) agree: the analysis methods that are used must be validated before being used routinely, and in the form that will be routinely used.
In other words, validating our control and measurement methods therefore serves to demonstrate to the authorities, via tangible evidence and objectives, that the techniques developed meet both the needs and requirements for which they were developed and that they give results that are reliable, and sound.
Why No. 2. Demonstrating our capacity to measure
Any analysis method, however effective it may be, is necessarily tainted by an error. All statisticians will tell you that we can never know the true value characteristic of a product or a process with certainty, but only come close to it. It is all a question of error and probability, which can be summarized in one word “uncertainty”.
Validating a method, from a statistical point of view, comes down to evaluating the total error inherent in the method and in comparing this with acceptance criteria that we can consider an acceptable error.By the total error of an analytical method, we mean the sum of the systematic error or bias of the method and of the random error or precision of the method, which are explained separately in the previous article in this journal (“Some Good Practices for Validating Analytical Procedures”). Validating a method is also to estimate the probability that, regardless of the day, time, technician, equipment, batch of reagent, etc. used to conduct the analysis, the results obtained will be fairly close to the real value which will remain unknown. In summary, validating an analytical method comes down to demonstrating a capacity to measure, quantify, qualify, characterise in a reliable and secure manner.
Why No. 3. Guaranteeing data integrity
In validating their analysis methods, laboratories evaluate their trueness and their precision; through this exercise, they also verify that the data generated are accurate and consistent, attributable to a given sample, legible, etc.
Validating analysis methods therefore allows laboratories to partially meet a second requirement of the health authorities: that of guaranteeing the integrity of their data at least partially, in accordance with the definitions provided in section 1 of this article.
NB: Validation is an essential step but is not enough – data integrity requires other elements, such as the securing of data items which involves validation of the information systems used to generate, process, store and archive them, among other things.
Why No. 4. Reducing the risks associated with our decision-taking
Today we have a multitude of information. We measure, collect, record, store and analyse a whole pile of data, but for what purpose? It is always the same: to reduce the risks associated with our decision-making.
In the context of healthcare industries, some of this decision-making is critical as it is liable to impact the health of a patient directly. It is therefore essential that the data on which these decisions are based are complete, reliable and secure; in other words that they are sound.
In conducting method validations and evaluating the total error of their analysis methods, laboratories increase confidence in the data on which they will rely for decisions such as the release of batches.
Why No. 5. Meeting the objectives of the health authorities
Guaranteeing the effectiveness, purity and quality of manufactured products, through reliable analysis.
5. The accuracy profile, an appropriate response to these “whys”
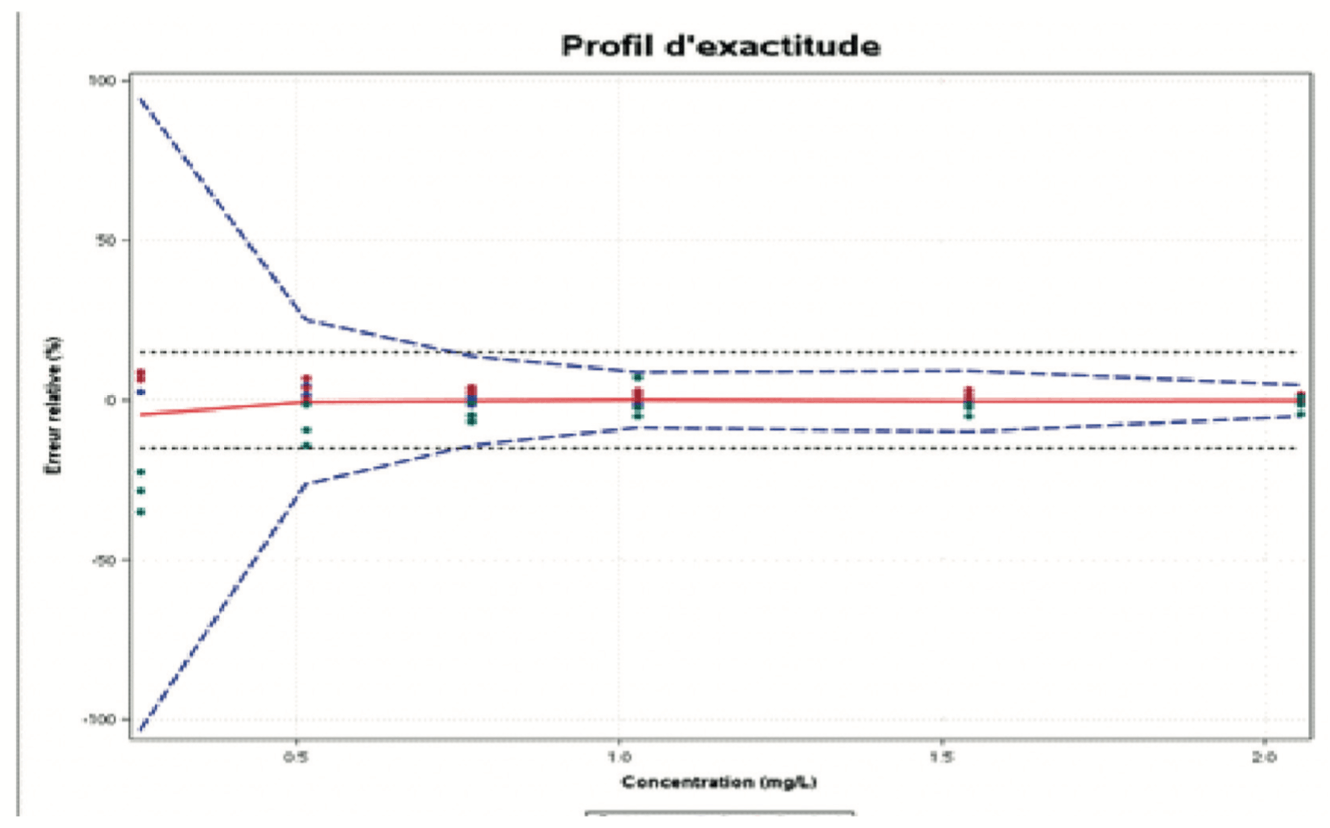
The accuracy profile is a useful statistical, graphic tool for the evaluation of the performance of an analytical procedure based on total error (bias + spread) and tolerance interval principles.
The role of the accuracy profile is to estimate, from the results obtained, what guarantee the user will have that the method will provide acceptable results when used routinely. It allows the approach to validating an analytical procedure to be simplified, while controlling the risk associated with its use. The use of an accuracy profile (Figure 3) involves fixing the following two decision criteria:
- The limits of acceptability of the method. These serve to translate the practical objectives of the users. They define an interval around the reference value. Most often, these limits are regulatory or derived from regulations. If there is no established reference, however, the expectations of end users (such as a given quantification limit), and the nature of the decisions to be taken on the basis of the measurement results should be taken into account.
- The BETA Proportion. This represents the proportion of future results that will fall within the tolerance intervals. The value chosen for BETA depends largely on the field of application (sanitary control, manufacturing control, etc.). It is evident that the lower the BETA value, for example 80%, the greater the risk that the method will produce results that do not correspond to the stated specifications.
Since it was introduced, although this tool is:
- very useful and effective for evaluating the true error of methods,
- applied,
- and recognized in different sectors of activity for all types of measurement methods and contexts (product or biological matrix, etc.),
several obstacles or reservations have been raised, to which answers can be provided.
a. There remains some confusion of terminology in certain sectors of activity, particularly between trueness and accuracy, or between reproducibility and intermediate precision…
And yet these definitions have been clear for 30 years. [10]
b. In some laboratories, total error causes annoyance because the accuracy profile shows a greater error than previously…
Yes but previously this was not altogether the true error of the method and validation is not merely a matter of statistical tests. It should be remembered that before the accuracy profile, applied statistics(ANOVA, STUDENT …) were giving rise to the following remarks in laboratories: “… the statistics are penalizing us…”, “…the better we work, the more we are penalized statistically…”, “… it’s difficult reaching a conclusion on the overall performance of the method with all these interlinked statistical tests….“
c. For some, tolerance interval calculations were complex…
But in the end, the interpretation of results is simpler.
d. Resistance to change, for example for the regulatory dossier aspects
Yes, but working with total error does not prevent each of these errors being presented in reports?
e. For some, the profiles are not obligatory in the dossiers
Yes, but they are very much the trend in AQbD, the future ICH Q14, the future USP 1220 or existing standards (USP 1210, V03-110 and T90-210, standards for QPCR methods, etc.) and this provides opportunities beyond actual requirements: it is a response to a user need to include the part potentially caused by analytics and that which is thought to be a manufacturing problem in the management of OOS.
However, we must remember that this profile was proposed initially in 2003 as it had been observed very regularly that validating a method using “dissociated errors” (bias and spread separately, and not the sum of the two errors) was leading to two recurring questions:
- How is it that we have so much difficulty in transferring this method when it had been declared VALID (…yes, but using “dissociated errors”)?
- How is it that we routinely have so many repeat assays/analyses?
Most of the arguments “against” are generated by a classic resistance to change. The authors of this article hope that point 5 will help the maximum number of people to take the plunge.
The contribution provided by an accuracy profile:
- Brings us closer to reality
- Facilitates analytical transfers
- Reduces repeat assays
- Allows us to implement a “RISK” management approach
- Allows us to run fewer statistical analyses
- Provides a simple interpretative tool
- Allows better control of the analytical procedure and its error
- Helps to estimate measurement uncertainties
- Reassures assessors, auditors, inspectors, customers, consumers
And therefore ultimately helps to improve data integrity (A for ACCURACY in the ALCOA principles)
Without going into the differences between the accuracy profile (with tolerance interval) explained here and the prediction interval described as an alternative in USP chapter <1210>, it is good to note that both steps are based on similar approaches. The recent arrival of the concept in the USP will democratize this type of approach that has been known in other scientific fields for more than 30 years.
We no longer have the desired reliability.
Conclusion
Regulatory requirements aside, each laboratory must obtain the strongest guarantees possible that the results generated are reliable.
The “total error” approach, which is representative of the unknown error with which every analyst is confronted during each batch analysis, answers the majority of questions that may arise when we address the role of validating analytical procedures in a broader manner.
And this “total error”, called Accuracy in ISO standards terminology, is indeed the same as one of the 2 As in ALCOA. Therefore, before thinking about the L, the C, the O, the other A and the + in ALCOA+, it is good to have already ensured the Accuracy of the data generated!
Share article


Eglantine Baudrillart – Neovix Biosciences
Eglantine est Responsable Business Developement du groupe neovix biosciences. Ingénieur en génie chimique (ENSIC) et diplômée de l’Advanced Management Programme de l’EDHEC BUSINESS SCHOOL, Eglantine a débuté sa carrière au sein des Laboratoires GSK par la mise en place d’un pôle de modélisation mathématique et statistiques puis a rejoint en 2008 les Laboratoires Expansciences au poste de responsable éco-conception procédés puis a repris la direction d’un laboratoire d’analyse accrédité.

Eric Chapuzet – Qualilab
Eric est Directeur Général de QUALILAB, société spécialisée dans le conseil en management de la qualité, sur le cycle de vie de méthodes d’analyse, les systèmes d’informations, l’intégrité des données et l’analyse de données. De formation scientifique (statistique, informatique, qualité, physiologie, pharmacologie, biochimie), il participe régulièrement à des projets de validation et transfert de méthodes analytiques. Éric anime et réalise également des formations et des audits dans le cadre des étapes du cycle de vie des méthodes d’analyse depuis de nombreuses années.

Gerald de Fontenay – Cebiphar
Depuis plus de 25 ans, Gérald a assuré différents postes au sein de sociétés de services analytiques et de CDMO. Aujourd’hui Directeur Scientifique et Technique chez Cebiphar, il continue à œuvrer pour garantir le maximum de fiabilité aux résultats qui sont générés dans ses laboratoires. Il apporte son expertise en validation, vérification et transfert de méthodes analytiques auprès des chefs de projets et des clients de Cebiphar, pour assurer le succès des projets qui lui sont confiés.
gdefontenay@cebiphar.com
Marc François Heude – Siegfried
Titulaire d’un doctorat en chimie organique, Marc assure, depuis 6 ans, des rôles de chef de projets analytiques d’abord à Adocia (Société de biotechnologie Lyonnaise) puis à Bayer Healthcare. Il exerce aujourd’hui en tant que responsable du service de développement analytique chez Siegfried, un CDMO de l’industrie pharmaceutique qui produit des APIs et intermédiaires de synthèse.
References
[1] : EudraLex – Volume 4 – Good Manufacturing Practice
(GMP) guidelines.
[2] : MHRA (Medicines and Healthcare Products and Regulatory Agency), «GxP Data Integrity Guidance and Definitions, 2018.
[3] : P. Hubert, J.-J. Nguyen-Huu, B. Boulanger, E. Chapuzet , P. Chiap, N. Cohen, P. Compagnon, W. Dewé, M. Feinberg et M. Lallier, «Validation des procédures analytiques quantitatives
harmonisation des démarches,» S.T.P. Pharma Pratiques, vol. 13, p. 101, 2003.
[4] : US Pharmacopoeia: USP <1210>.
[5] : Pharmacopeial Forum (PF), 1220 Analytical Procedure Life Cycle — PF 46(5), 2020.
[6] : FDA Guidance: “Analytical Procedures and Methods Validation for Drugs and Biologics”, 2015.
[7] : ICH Q2 (R1) “Validation of Analytical Procedures: Text and
Methodology”, 2005.
[8] : G. de Fontenay, «”Replicate Strategy” : mais quel est ce concept ?,» La Vague, vol. 68, p. 34, 2021.
[9] : US Pharmacopoeia: USP <1210>; <1225>; <1226>. [10] : NF ISO 5725. Application de la statistique. Fidélité des méthodes d’essai. Détermination de la répétabilité et de la reproductibilité d’une méthode d’essai normalisée par essais interlaboratoires., 1987.
