

Rationaliser et prioriser les dispositions de maîtrise de la fabrication d’un médicament : tel est l’objectif du Quality Risk Management (QRM). Bien que sur le fond, l’approche par le risque soit une méthodologie utilisée et exploitée depuis longtemps dans d’autres domaines industriels, pour les entreprises pharmaceutiques, du fait de leur mission de santé publique, la mise en oeuvre est beaucoup plus délicate.
L’approche QRM est rendue efficace et pertinente par son intégration avec la démarche Quality by Design, qui place la connaissance du produit et la compréhension du procédé au coeur des décisions. Repositionné progressivement depuis 2005 par les autorités réglementaires comme fondement de la qualité, le Quality Risk Management (ou gestion des risques qualité) repose sur deux fondamentaux : une évaluation du risque basée sur la connaissance scientifique et un effort de formalisation et de documentation qui doit être proportionné au risque.
Un enjeu pour les Biotechnologies, un challenge pour les petites structures
De par la complexité des produits issus du vivant, l’évaluation et la priorisation par le risque sont déterminantes dans le cadre des activités de développement et de mise à disposition de médicaments issus des biotechnologies. Elles permettent, en effet, de prioriser les études, de piloter et communiquer le niveau de maîtrise du risque patient tout au long du cycle de vie du produit, et ce, en ligne avec les attentes des autorités. L’enjeu pour les laboratoires de biotechnologie est d’augmenter de façon organisée et ciblée la compréhension et la connaissance de leur produit par la mise en oeuvre du QRM. L’approche Quality by Design, telle que définie par les guides ICH Q8 et Q11, propose notamment de structurer la connaissance pour assurer le lien vers la sécurité du patient tout au long du développement. A chaque stade, la gestion du risque permet d’éviter de découvrir, trop tard, des sources de variabilité non maîtrisées, avec les impacts de retard sur le planning qui sont associés.
Qu’est-ce que la gestion du risque qualité ?
Un risque est rattaché à une situation de risque ou un aléa et se caractérise par la combinaison de sa gravité et de sa probabilité. Le Quality Risk Management tel que proposé par l’ICH Q9 se décline en quatre étapes: l’évaluation du risque (1), la maîtrise du risque (2), la communication du risque (3) et la revue du risque (4). (Fig.1)
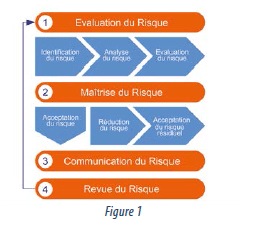
La première étape consiste en l’identification de la situation de risque, l’analyse du risque par l’application de tables de cotation (gravité et probabilité) et l’évaluation du risque pour déterminer le niveau de risque associé à l’activité ou l’opération réalisée. Sur la base des résultats de l’évaluation, la seconde étape d’analyse est initiée par le choix d’accepter ou non le niveau de risque obtenu en regard de seuils de décision préfixés. S’il est considéré comme non acceptable, il convient de définir des moyens de réduction du risque jusqu’à le rendre acceptable, par exemple en diminuant la probabilité de survenance de la situation à risque ou en augmentant la détectabilité sur le produit lui-même ou sur les incidents à l’origine d’une non-conformité. Pour ce faire, la mise en place de contrôles additionnels est généralement réalisée (procédure, tests analytiques, validation, traçabilité…).
Cette étape de maîtrise permet de justifier et rationaliser les efforts entrepris et donc de légitimer les choix stratégiques adoptés. Le risque accepté fera l’objet d’un engagement formel.
La troisième étape permet la communication des risques et leur prise en considération par les acteurs pharmaceutiques.
Enfin, la quatrième étape permet la mise à jour des dossiers de gestion des risques pour prendre en compte de nouvelles connaissances ou des évolutions suite aux retours d’expérience, susceptibles d’impacter la stratégie de maîtrise de risques établie lors de l’exercice QRM. Le dossier de risques se consolide ainsi par itérations sur l’ensemble du cycle de vie du produit.
La mise en application dans les phases du développement
Pour une mise en application efficiente, pour chaque étape, il est important de bien considérer l’objectif et le périmètre d’application pour ne pas se lancer dans des analyses inutiles ou trop chronophages au vu de l’enjeu à considérer (2).
Quelques cas d’étude appliqués aux biotechnologies ont été publiés et permettent d’illustrer une approche couplant les activités de développement et la prise en compte du risque (3).
Le point de départ reste le Profil Qualité Cible du Produit (Quality Target Product Profil / QTPP) à partir duquel sont définies les caractéristiques attendues du produit (Product Quality Attribute / PQA).
Si une dérive de ces attributs impacte la sécurité ou l’efficacité du produit, ils deviennent par définition des Attributs Qualité Critiques (Critical Quality Attributes / CQA). L’évaluation par le risque de ces CQA permet un classement (ranking) des éléments les uns par rapport aux autres, permettant de prioriser les études de développement à mener. Par exemple, les seuils toxicologiques de certaines substances, les effets cliniques connus du produit ou simplement l’absence de connaissance suffisante pour caractériser l’impact sont des critères qui permettront de pondérer le niveau de risque.
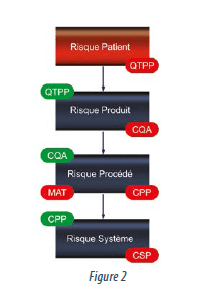
Le procédé de fabrication est développé pour permettre l’obtention in fine des PQA du produit mais avec une priorisation sur les CQA, pour conclure rapidement au possible lancement ou non de la molécule.
Les liens qui couplent une qualité de matière entrante (Material Attribute / MAT) à des paramètres procédé pour obtenir la qualité de matière sortante voulue sont les clés issues de la compréhension du procédé. C’est par un enchaînement d’étapes de transformation de la matière, comme illustré ci-dessus (fig.2), que l’on aboutit au produit fini. Un paramètre procédé impactant un MAT lié à un CQA est critique (Paramètre Procédé Critique ou Critical Process Parameter / CPP). Dans cette analyse, le risque intervient pour définir les études de robustesse et les plans d’expérience à mener afin de caractériser les manques de connaissance, ou d’affiner les conditions opératoires de mise en oeuvre. L’évaluation par le risque permet de justifier des paramètres critiques bien maîtrisés (Well controlled CPP / WC-CPP). En effet, certaines caractéristiques produit, bien qu’ayant un impact fort sur le patient, ne nécessitent pas nécessairement d’études spécifiques poussées, car leur mise sous contrôle est bien connue (Stérilité, Formulation…). Toute la prise en compte du système de production à l’échelle industrielle est réalisée dans les étapes finales du développement et dans le transfert.
Quelle mise en place dans l’entreprise ?
Comme défini par ses fondamentaux, la gestion de risque est basée sur la connaissance. Il y a un risque à dépenser du temps et des efforts pour constituer une connaissance si elle n’est pas indispensable à la maîtrise de la sécurité patient.
De fait, pour être efficace, une approche par le risque doit être portée par les métiers concernés, tout en étant soutenue par un garant méthodologique permettant une bonne compréhension de la méthode par les experts, par une organisation robuste et par le senior management, pour mobiliser les bonnes ressources, et aligner l’ensemble des acteurs vers la gestion de la qualité en prenant en compte le risque patient. Elle nécessite également la mise en place d’une équipe pluridisciplinaire pour intégrer toutes les composantes du risque. Lors de sa mise en oeuvre, l’approche par le risque doit être gérée comme un vrai changement culturel pour en tirer tous les bénéfices envisagés.
Quel investissement ?
Le lancement d’un changement de culture relève d’une politique d’entreprise.
En regard de l’organisation des grands groupes pharmaceutiques, la plupart des sociétés de biotechnologie ont intérêt à profiter de leur agilité et de leur réactivité pour s’orienter vers des solutions adaptées non systématiques, une politique globale, la conduite du changement et la mise à disposition d’outils pratiques. L’objectif principal est bien la mise en place d’une réflexion par le risque, évolutive au fur et à mesure de l’avancement des projets, en profitant si possible du retour d’expérience de personnel tiers (soustraitant, fournisseur, client et/ou partenaire), avant d’homogénéiser et standardiser les pratiques.
Un travail préparatoire est à budgéter pour transposer les fondamentaux dans le contexte de l’entreprise, fixer la politique, mettre en place l’organisation qui va supporter l’évolution vers l’approche par le risque. Bien évidemment, il s’agit de fixer des responsabilités, monter et piloter des plans d’actions, et pas nécessairement de créer des postes !
Pourquoi aller et quand aller vers le QRM ?
Aujourd’hui, tous les dossiers d’enregistrement déposés au niveau de l’EMA ou de la FDA sont évalués avec une vision Quality by Design / Risque. La mise en place d’une approche par le risque devient donc indispensable pour tout nouveau produit et conduira à des évaluations, audits et inspections facilités.
Par ailleurs, cette gestion par le risque stimule la mise en place de la gestion de la connaissance (Knowledge Management / KM) et soutient le maintien et le développement des connaissances scientifiques par les laboratoires. Enfin, l’approche par le risque fournit une réelle clé d’investigation et d’évaluation pour les différents processus qualité (CAPA, Change, Déviation…).
Les gains apportés par la mise en oeuvre du QRM sont générés principalement par des soumissions alignées aux attentes de l’évaluateur (démarche «Right First Time»), et par l’accélération et la fiabilisation du traitement des événements pour les productions de routine. La réduction des délais d’enregistrement et des coûts de la non-qualité peuvent aisément s’élever à des millions d’euros.
En complément, le nouveau guide ICH Q12 aura pour objectif de définir une nouvelle approche en matière de gestion des changements post-approbation, impulsant l’intégration du Quality by Design (QbD) dans la phase d’exploitation commerciale du cycle de vie du produit.
Pour cela, l’implémentation du QRM est un changement de culture à conduire, en pilotant l’évolution de la maturité de tout le système Qualité et ses acteurs en terme de gestion par le risque patient.
Comme tout changement culturel, il doit s’appuyer sur une définition claire de l’organisation des responsabilités et être soutenu par des messages fondamentaux et par une communication cohérente afin que les autorités elles-mêmes ne considèrent pas le manque de maturité « risque » comme inacceptable.
Bibliographie :
1 – a) ICH Q9 – 2005 b) Japan : Ministry of health labour and welfare MHLW : Product GMP Guidelines “Annex” c) EU-EMA : GMP Annex 20 – Quality Risk Management – février 2008 d) EU-EMA : GMP chap I – Quality Management System – Mars 2013 e) US-FDA Guidance for Industry – Process Validation: General Principles and Practices – Janvier 2011 f) EU-EMA Guideline on process validation for finished products – information and data to be provided in regulatory submissions – Janvier 2014 g) EU-EMA GMP annex 15 Qualification & Validation – Mars 2015
2 – PDA TR 54 : Implementation of QRM for Pharmaceutical and Biotechnology Manufacturing Operations (2012)
3 – A-VAX : Applying Quality by Design to Vaccines – CMC Biotech Working Group – version 1.0 (Mai-2012)
Camille Landrieu – AKTEHOM
Priscille PINGAULT – AKTEHOM
 |
| L’article en version PDF |
