


Sommaire
- Validation Strategy of Viral Decontamination Methods, a quick overview
- Antibody-Drug conjugate Manufacturing Techniques
- Robust and Convenient Single-use Processing
- Cahier Pratique – Quality by design applied to viral safety of Biologicals: Case studies & workshop discussion summary
- Mass spectrometry as a powerful tool for the characterisation of monoclonal antibodies in the context of comparability studies
- Chromatographie Continue : Solution d’amélioration des performances de procédés et « debottlenecking » des capacités de Bioproduction
- Protein A Affinity Chromatography for Efficient Fab Purification
- Enabling Higher Post Protein A Product Purity Using Novel Chromatographic Clarification Approach
Applying Quality by Design (QbD) to Viral Safety of biologicals has started to be discussed and implemented to evaluate the robustness of the viral clearance of processes while providing a design space where variations can be acceptable. While numerous QbD studies have been initiated to assess the process performances, limited publications are available on QbD application to viral safety. In this article we use examples of virus filtration and chromatography to discuss how knowledge base and industry experience can help design a risk assessment and a QbD approach for virus removal steps in a biological process.
This article summarize case studies discussed in the workshop the A3P congress of Lausanne in April 2015. This workshop was aiming at introducing the concept of QbD applied at viral safety and discussing real cases and data from LFB Biotechnologies and Merck.

The Quality by Design (QbD) approach has started to be implemented in the biotech industry to ensure the quality of biopharmaceuticals. This initiative allows a thorough understanding of the drug process and the design of a space where variations can occur without altering the quality and the efficacy of the final product. Once validated and approved, the QbD approach may allow more flexibility in manufacturing process changes and reduce change control burden. The concept foundations lay in the International Conference on Harmonization of technical requirement of Pharmaceuticals for Human Use (ICH) guidelines, Q8R2 (Pharmaceutical development), Q9 (Quality Risk Management), Q10 (Pharmaceutical Quality System) and ICH Q11 (development and manufacture of drug substances (chemical entities and biotechnological/biological entities)).
The QbD concept is a stepwise approach and the initial task is to define Critical Quality Attributes (CQA) of the final product, which characterize these attributes and their impact on the product’s quality, safety and efficacy (Figure 1). Typically this identification is a result of a thorough risk analysis (according to ICH Q9) and scientific characterization of the product and process. Once the CQAs are identified, the next step is to outline the work flow that needs to be developed in order to ensure the process will manufacture product that meets its product attributes objectives, as defined by the CQAs and eventually the quality target product profile (QTPP). The development of this workflow involves the definition of a design space, a multidimensional area where process parameters can vary and interact in a multivariate fashion. Finally, a control strategy will use process controls on critical process parameters (CPPs) to constrain operations to design space whilst still having acceptable CQAs. The clear advantage of the characterization of this design space is to allow the manufacturer to deal with changes internally and alleviate regulatory approval burden, providing that changes are made within the already validated and approved design space.
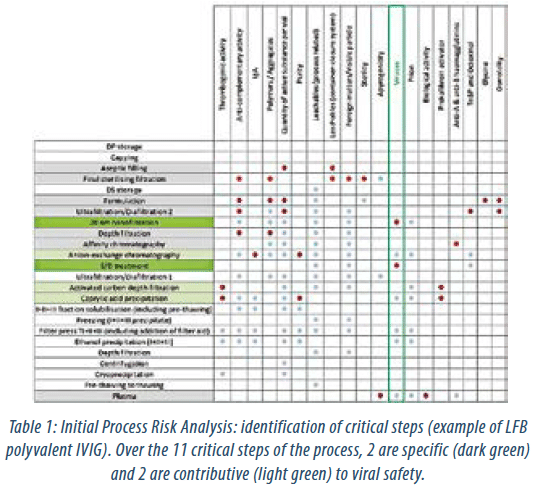
A process risk assessment is an essential and very effective way to connect the product design, process unit operations and final product performance CQAs. The first step is to evaluate each process step against the defined CQAs in order to identify process steps that would require further characterization. Figure 2 shows an example of the identification process of critical steps for LFB polyvalent IVIG for viral safety of the product, as this is considered an important CQA. The second step is focused upon the potential impact of the process parameters. Establishing a process-design space starts with the definition of CPPs that are likely to have a significant impact on the CQAs and therefore on the quality, safety or efficacy of the product. Any parameter identified having a high potential impact on CQAs is therefore targeted for further study (Table 1). This is accomplished by accumulated knowledge of each step including inhouse process development data and from suitable literature. The capacity of a step to inactivate and/or remove viruses remains an important aspect to achieve an acceptable viral safety profile.
| Critical Steps | Contributive studies |
| Caprylic acid fractionation | Process robustness study |
| Activated carbon depth filtration | Viral clearance study |
| S/D treatment | Viral clearance study |
| Anion-exchange chromatography | Process robustness study Viral clearance study Ageing study |
| Affinity chromatography | Process robustness study Ageing study |
| Depth filtration | Process robustness study |
| Virus filtration (nanofiltration) | Viral clearance study |
| Ultrafiltration/Diafiltration 2 | Process robustness study |
| Formulation | Validation batches |
| Final sterile filtration | Sterile filtration validation study |
| Aseptic filling | Filling validation study |
| DP storage | Stability study |
Table 2: Process Risk Mitigation Studies (example of LFB polyvalent IVIG)
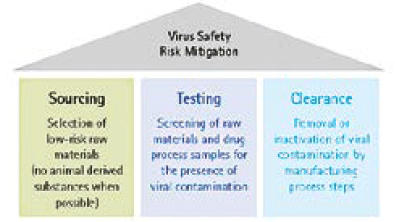
The assurance and calculation of appropriate viral clearance is an important CQA and should be considered for all products that derived from mammalian cell culture or from biological sources such as monoclonal antibodies, recombinants, vaccines and plasma derived proteins. In the case of products that derive from mammalian cell culture, the rationale of such consideration is first that mammalian cell cultures are known to produce endogenous retroviruses or retrovirus-like particles (RVLP) and second that they have the potential to be contaminated by adventitious viruses during the production or from animal derived raw materials. Blood derived products can carry contamination from the original donors. Regulatory guidelines stress the importance of a multi-layered orthogonal strategy to ensure viral safety of biologicals, which include the
1) The selection of low risk source materials,
2) Appropriate testing and
3) Implementation of viral inactivation and removal steps (Figure 2).
Applying QbD in the selection and testing of raw materials (1) and in the viral removal or inactivation steps within the manufacturing process can provide validated assurance of an effective and consistent overall viral clearance strategy.
One can understand that such approach implies a level of investment in the development phase. To date there are limited examples in the literature that describes an approach CQA selection and design space generation dedicated to viral clearance. Genentech (a Roche company) reported the development of viral clearance steps of monoclonal antibodies processes where QbD was applied, specifically on Protein A and anion exchange (AEX) chromatography and on viral filtration (3). They used design-of-experiments (DoE) and found that simultaneously varying multiple process parameters did not affect the ability of the AEX process to remove endogenous RVLPs or virus models such as xMuLV, MMV (mouse minute virus) and SV40. For Protein A they also did not find that varying process parameters had significant effects thus allowing the establishment of a large design space. Similar findings were reported for viral filtration. Nevertheless, they have observed difference between feedstocks on the AEX and the impact of the loading amount on virus filtration Log Retention Values (LRVs).
Multiple unit operations will typically contribute to the overall viral clearance, i.e. an amount of log reduction value (LRV) acceptable to ensure the safety of the final product. The workshop “QbD approach applied to viral safety of biological” of the A3 Conference, in Lausanne, in April – focused on the QbD in Viral clearance, specifically chromatography and viral filtration(4). The objectives of the workshop was to open the discussion on the choice of critical parameters for these unit operations and brainstorm on the approach and methodologies that would support the formation of the design space whilst optimizing resources and time.
Viral clearance studies are costly (typically performed at a contract research laboratory) and time consuming. A full and extensive product and process characterization would be required to define a viral safety design space and this might not be practical with the ressources available. A strategy of establishing a safe space based on the science, risk based selection of parameters (past experience, process step mechanism of viral clearance) and experimental data is rather applicable. Several points that need to be taken into account whilst designing a viral safety study were discussed during the workshop:
| Main steps | Contributive steps |
| Solvent/detergent or detergent treatment | Chromatography (anion-exchange, cation-exchange, affinity) |
| Virus filtration (nanofiltration) | Fractionation (ethanol, caprylic acid) |
| Low pH incubation | |
| Dry heat | |
| Pasteurization |
Table 3: Viral clearance steps in biological processes
1. Choice of the process step: specific and/or contributive step to virus removal
As mentioned earlier, there are few examples available in the literature of QbD approach applied to viral safety that could allow a clear selection process to be drawn. Validation studies as guided by regulatory documents typically cover a minimum of two different steps to comply with the orthogonal approach requirement. Additional contributions can come from other steps in the process but are not necessary covered by the validation studies, unless the overall LRV achieved is insufficient to guarantee an acceptable safety assurance for marketing authorization. In a biological process, it is considered that viral filtration (5) and viral inactivation (e.g. low pH incubation, Solvent/detergent) – when applicable – are the main viral clearance steps(6) (Table 3). Chromatography steps are also routinely tested for their ability to remove viruses but are generally considered contributive. AEX has been well established as robust and predictable step for removal of viruses including retroviruses and parvoviruses (≥ 4 LRV)(7). In monoclonal antibodies processes, Protein A has been shown to provide virus reduction capabilities but is generally considered less effective than AEX (8). Cation exchange chromatography (CEX) is used as a polishing step in some biological processes. The ability of CEX to contribute to virus clearance has been studied by various groups and findings were contradictory. Regardless of the study, it is accepted that a high number of variables directly and indirectly influence viral clearance by CEX including operating pH, elution salt concentration, ionic strength of the equilibration and loading buffer, impurity level, etc (9). Altogether this knowledge base provides a focus for the QbD approach on the steps that are considered the most effective and typically subjected to virus validation studies for clinical material. For the purpose of the workshop, virus filtration (one main step) and AEX (one contributive step) were selected for further discussion.
2. Choice of Critical Process Parameters (CPPs) for anion exchange chromatography & viral filtration
For AEX, previous studies have demonstrated that feedstock parameters such as pH and conductivity can have an impact on LRV values and can interact with one another in a multivariate fashion(10). Impurity levels (HCP, DNA) can have a strong impact on virus removal especially at higher distribution coefficient and higher load challenge (11). It is critical to control the feedstream impurity levels to avoid impurity breakthrough and competitive binding in one hand and assess the impact of residual impurities on virus removal via chromatography. Virus removal of the AEX is generally considered less sensitive to contact time (bed height), flow-rate, resin life time and pooling methods(12). Finally intrinsic product and virus characteristics such as pI need to be taken into consideration when designing DoE experiments.
For viral filtration, the change in pressure or the interaction of pH and pressure has been found in certain cases to affect the LRV value (13). This observation has been restricted to only some of the commercially available parvovirus retentive filters, highlighting that a given parameter or the interaction of multiple parameters do not equally impact the virus filters available in the market. Therefore, including these variables in early studies is recommended, especially when filters from different manufacturers are implemented for one unit operation to enhance security of supply. In a recent study, the commercial parvovirus retentive (14) filter Viresolve® Pro was further evaluated for its parvovirus retention after process interruption where forward pressure was released (15). Parameters such as timing of the pause, duration of the pause, pore size distribution of the filter membrane and presence of the product were assessed. No significant LRV changes were observed, confirming the initial observations in the previous studies aforementioned. Bolton et al (16) have observed a relationship between fouling of normal flow virus filter and LRV decline. Using experimental and mathematical models, they reported that the mechanism of LRV decline was due to selective plugging of small pores. As protein solutions may vary in their behavior during filtration the evaluation of the LRV vs flow decay relationship was recommended. Following this observation, studies were performed to assess feed load and mass throughput on commercial filters. An example is provided in the table 4.
| Load (g/L) | Mass throughput | LRV at 50% target volumetric throughput | Final % flow decay | Final pool LRV |
| 2 | 2.0 | N/A | 93 | ≥5.8 |
| 7 | 8.4 | ≥6.1 | 86 | 5.8 |
| 15 | 14.6 | ≥5.8 | 87 | ≥5.8 |
| 15 | 16.3 | 5.3 | 89 | 4.8 |
| 25 | 20.2 | 5.4 | 88 | 5.7 |
| 25 | 20.4 | ≥6.0 | 90 | ≥6.0 |
Table 4: Assessment of a mAb feed concentration variation on MVM retention of a commercial filter, Viresolve® Pro(17)
Feeds were supplied at 2, 7, 15 and 25 g/L solutions in a phosphate buffer. Before testing feeds were 0.22 μm filtered then prefiltered prior to addition of virus. Devices were challenged at 2 bar with protein solutions spiked with MMV to a target titer of 2 x 106 TCID50/mL. Samples were collected from the filtrate pools at designated volumetric throughput and samples were assayed for infectivity using the standard cell based TCID50 assays. Protein levels were measured in all samples by OD280 readings.
All devices were stopped after reaching 80–90% flow decay (approximately four hours after initiation of the test). Effective clearance of MMV was observed across all concentrations with LRVs greater than 4.8, indicating that retention by Viresolve® Pro Devices is not impacted by protein concentration.
The impact of conductivity on virus filtration has also been assessed by different groups. While in some situations it was found that LRV was not impacted by the variation of ionic strength(18) , it was observed that certain ionic strengths were associated to LRV decrease (19).
Filter Shelf life & lot to lot variability are generally assessed to demonstrate the technology robustness and ensure process consistency. Viresolve® Pro retention was assessed across the time using different devices and Phage X174 (Table 5). The study result demonstrated that shelf life was not influencing retention capabilities of the filter. Similar studies were performed to assess lot to lot consistency and results were different across filter suppliers (20).
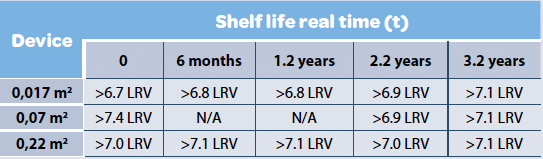
Viresolve® Pro Modus devices (0.017, 0.07 & 0.22 m2 ) were spiked with Phage X174 in 50 mM sodium acetate, 100 mM NaCl, pH 5.0, at flow rate of approximatively 10 L/m2 at 30 psi and TCID50 values were recorded for each experiment. N/A indicate absence of data.
Table 6 is a summary of proposed parameters to include in a QbD approach for AEX & virus filtration unit operation development.
| Key Design Space Parameters | |
| AEX chromatography | Virus filtration |
| Feedstock pH | Feedstock pH |
| Conductivity (mS/cm) | Conductivity (mS/cm) |
| Buffer pH | Pressure & process interruption (Bar or Psi) |
| Impurity level | Load (L/m2 or Kg/m2) |
| Product concentration (load density g/L) | Product concentration (g/L) |
| Flow rate (CV/hr) | Flow decay (% plugging) |
| Column volumes (Cm/h, elution, washing, loading) | Filter shelf life |
| Temperature °C | Filter lot |
Table 6: Proposed parameters to include in the risk assessment phase
3. Univariate or multivariate studies ?
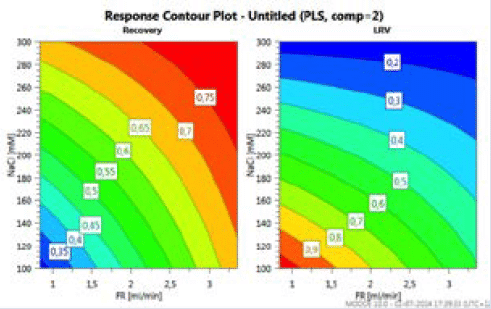
During the workshop, the rationale behind conducting univariate or multivariate studies was discussed. A univariate study performed by Genentech was reported and was restricted the protein load (21). This approach was more considered for business risk mitigation rather than for the viral safety risk. It was proposed that multivariate DoE studies allow to assess the interaction of some parameters and allow the reduction in the number of studies. In a recent collaborative work between Merck and IBET to develop an insect cell culture derived virus-like particle vaccine process, AEX chromatography resins were evaluated for the removal of residual impurities including baculoviruses (22). DOEs were conducted while varying two parameters: salt concentration and flow rate to assess the VLPs recovery vs Baculovirus removal. The study showed an interaction between the two parameters and allowed to the identification of the best conditions to achieve desired recovery/removal ratio (Figure 3).
An LFB case study was presented where the effect of two factors (protein load and pressure applied to the filter) was evaluated on filter parvovirus removal (Figure 4). It was postulated that these two parameters could impact virus removal during the parvovirus filtration step as mentioned in the previous paragraph. These potential critical process parameters (CPPs) were therefore retained for following studies. The protein load was demonstrated not to impact on virus removal. In contrast, a trend toward lower log reduction factor was observed at lower pressures (arrow in the figure 4), suggesting an impact of lower pressures on filter parvovirus removal. Process limits have been, therefore, adjusted accordingly.
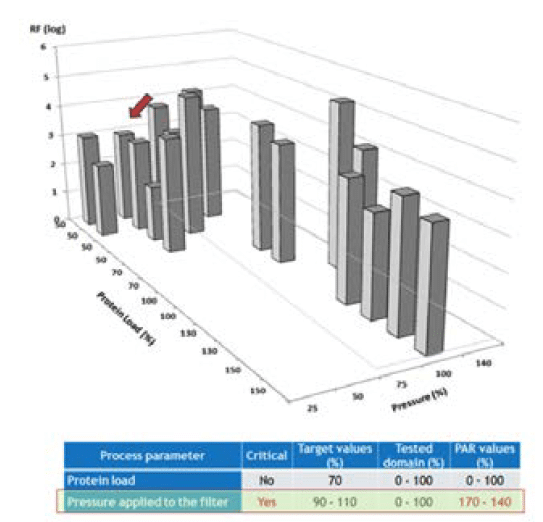
and 2 parameters modified in combination
Accomplishing multivariate studies requires a significantly greater upfront investment in development than does the traditional approach. However, the benefits include operational flexibility, as future modifications to the purification method that remain within the process design space are considered non-notifiable changes and do not require additional viral clearance studies. In addition, the impact of excursions during a production run can be confidently evaluated, reducing clinical and financial risk.
4. Virus models
The choice of virus models for virus removal validation studies derive from regulatory guidelines and common science and relates to the nature of the process. For instance, monoclonal antibodies are typically produced from CHO cell cultures, hence the choice of models such as MMV and xMuLV for the validation of virus removal. For processes that derive from human plasma, blood or urine, or cell culture derived vaccines, other relevant models are used. While the rationale of such choice is clear and understood for virus validation studies, their usage becomes a limitation factor for large DoE and QbD studies. Indeed, not all manufacturers have dedicated laboratories or personnel and outsourcing such studies can be costly.
In their study on AEX chromatography, Genentech proposed to use RVLPs for extensive DoEs. The preparation and the manipulation of RVLPs is less cumbersome and constrained than virus models like MMV and xMuLV. For the same reason, most filter manufacturers use phage models to characterize their virus filters. These models, allow more extensive studies and are relatively economic. The outcome of the workshop was that RVLPs or phage could be used for a first set of DoE studies. Subsequently depending on the result of these studies, a second set of smaller amount of experiments could be run with models that are accepted by the regulatory authorities.
5. Quality status of the studies: Should the studies follow Good Laboratory Practice (GLP) or Good Manufacturing Practice (GMP) or others?
Viral clearance studies are most of the time performed by a Contract Research laboratory and follow the principles of Good Laboratory Practice (GLP). Viral clearance robustness or QbD studies applied to viral safety are typically carried out at later stage of the product lifecycle, especially for marketing authorization. At this stage, a fully GMP-compliant validated production process is in place, viral safety studies performed at this stage should be therefore GMPcompliant. However, the cost of viral clearance studies being high, their feasibility within a QbD approach might be limited for some companies. Other strategies can be put in place to overcome this situation. During the workshop, it was discussed that one option could be to run the scaled-down viral safety QbD studies (spiking and sample generation) under an internal quality system (“GMPlike”) and samples can be titrated either under internal quality system approved assay (Polymerase Chain Reaction (PCR)) or at a contract lab (infectivity (TCID50)). Another option could be the generation of data (screening) with a nonhazardous, non-mammalian virus models (i.e. bacteriophages). The use of bacteriophages allow the generation of a large amount of data with relative speed and costeffectiveness and this can be performed under GLP or non-GLP conditions either in internal lab or at the supplier of the support (filter, resin, etc.). Ultimately, the approach should be recommended and accepted by the regulatory approval entities taking into considerations the points aforementioned.
6. Statistical methodologies and tools for interpreting large amount of data
The final step of a QbD study is the interpretation of the large amount of data, and this process is not trivial. Indeed, appropriate statistical support or software should be available to provide a structured and organized method to determine the relationship between factors (CPPs) and the outcome (LRVs). This appropriate statistical support should also provide confidence on the interpretation. The question during the workshop was if there is available software which provides confidence on the interpretation of LRVs.
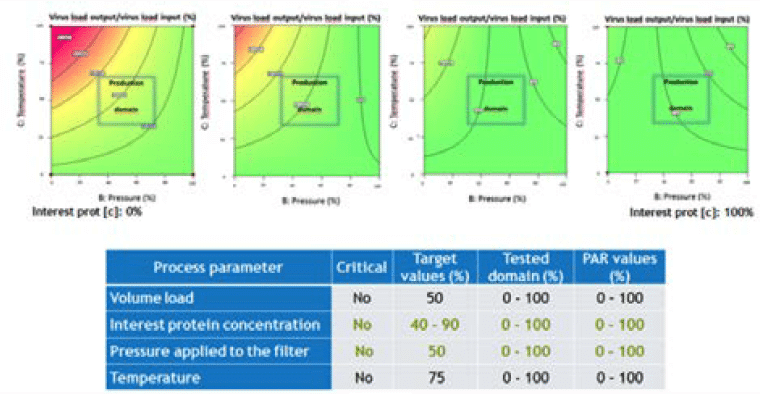
A second LFB case study assessed the effect of four factors (load volume, protein of interest concentration, pressure applied to the filter and temperature) on parvovirus removal (Figure 5).
This case highlighted the practical difficulties in interpreting the data, especially due to the use of two statistical methods (Poisson distribution when no virus is detected and Spearman Kärber method when virus is detected). Indeed, two factors seem to have a very minimal impact on virus removal (lower protein feed concentration and lower pressure applied to the filter). This minimal impact is mainly due to detection of virus particles; however, the log reduction factors obtained in these conditions are not statistically significant and very close to the value of the standard condition. It was therefore concluded that the process parameters are noncritical on the tested domain.
7. Group discussion outcome: QbD application to virus filtration study design
Final workshop discussions focused on viral filtration process step. Table 7 provided hereafter is a summarized proposal to help design and support QbD for virus removal filters.
| Pathogen agent | A first screening of the parameters with a phage, then selected critical parameters with a parvovirus |
| Selected parameter | Protein load, load volume, filtration time, pressure, process interruption selected as the main critical parameters. Temperature, pH, concentration, filter lot (data from the supplier) considered less critical |
| Univariate or multivariate | Multivariate (max. 3 parameters in combination) |
| Number of runs | Duplicate run for center point and single run for the other points. |
| Virus titration assay | Infectivity (TCID50) |
| Quality status of the assay | Non-GLP with phage in a first stage and GLP with parvovirus with identified CCPs |
| Statistical method | Convenient software needed |
Table 7: Summary of the group discussion related to parvovirus retentive filter design studies
Conclusion and perspectives
The industry is embracing the QbD approach and its application to virus safety is emerging. Upfront work and solid filtration knowledge is needed to understand how variations in-processing parameters impact the capacity of the manufacturing process to remove or inactivate a potential contamination and thus to define a proven design space. Knowledge base and validated work can allow narrowing down the exploration area. The role of the filter or chromatography resin (or any other technology) supplier is considered key as he can provide robustness studies that help this exercise. Through this approach the risk can be managed effectively and viral safety can be achieved. Still, the workshop outcome highlighted the difficulty to initiate large DoEs and the selection of relevant parameters and combinations as most of the recent works were based on historical experiences and knowledge bases. Moreover, using economical models might be a solution to alleviate the cost of such investigational studies. Finally, because such QbD approach has to be approved by regulatory bodies, more guidance will be needed in the respect of the application of QbD to viral safety to follow common procedures and standards.

Caroline GOUSSEN – LFB BIOTECHNOLOGIES
goussenc@lfb.fr

Anissa BOUMLIC – MERCK
anissa.boumlic@merckgroup.com
Partager l’article
Bibliographie
(1) Further reading: Kathryn Martin Remington, PhD. Application of Quality by Design to Viral Safety, BioPharm International, October 2014
(2) Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin Q5A – ICH Harmonised Tripartite Guideline
(3) Strauss et al, Biotechnol. Prog. 26, 2010 ; Zhang et al; Biotechol. Bioeng (2014)
(4) Also referred as nanofiltration
(5) Also referred as Nanofiltration
(6) Dichtelmüller et al, Transfusion 2009
(7) Curtis et al, Biotechnol Bioeng, 2003, Miesegaes et al, Biotechnol Bioeng 2010.
(8) Zhang et al, Biotechnol Bioeng, 2009
(9) Connell-Crowley et al, Biotechnol Bioeng, 2011
(10) Strauss et al, Biotechol Bioeng, 2009 ; Curtis et al, Biotechnol Bioeng (2003)
(11) Iskra et al, Biotechnol. Prog., 2015
(12) Strauss et al, Biotechol Bioeng, 2009; Curtis et al, Biotechnol Bioeng (2003); Norling et al, J Chromatography A, 2005
(13) Tomoko Hongo-Hirasaki, PDA Viral Safety for Biologicals, 2015 ; Lacasse et al, Bioprocess International, 2013
(14) Also refered as small virus retentive filter
(15) Leahy et al, 2015 PDA Virus/TSE Safety
(16) Bolton et al, Biotechnol Appl. Biochem, 2005
(17) Viresolve Pro® performance guide RF1013EN00, page 8
(18) Jorba N., et al., Virus Removal Capacity of Viresolve® Pro Filter during Alphanine® SD Nanofiltration Under Different Ionic Strength Conditions, PDA Virus/TSE, Conference Barcelona June 2011
(19) Tomoko Hongo-Hirasaki, PDA Viral Safety for Biologicals, 2015
(20) Dayue Chen, “Systematic evaluation of Virus removal by parvovirus nanofilters”, Biopharmaceutical Development & Production week, Carlsbad, California, 2010
(21) Christian Bell, Viral Safety for Biological conference, Cologne, 2014
(22) Pryabrata Pattnaik, Production and purification of a VLP based Hepatitis C vaccine candidate, 2015 PDA Europe Vaccine conference; Silva et al, ECI Vaccine Technology V Conference, Mexico, 2014.
