


Octobre 2024
La Vague n°83
EU GMP Annex 1 / Maitrise du risque patient ICH Q9(R1) / Digitalisation
Sommaire
- What does 21 CFR Part 11 mean in everyday online analytics?
- Digitalization in cleaning validation an overview of possibilities, challenges, and opportunities for savings?
- EU GMP Annexe 1. Mise en œuvre de la Stratégie de Contrôle de la Contamination
- Impact of the new Annex 1 on Sterile Filling
- The Challenges of Floor Cleaning & Sanitization
- Réduction énergétique des centrales de traitement d’air : comment adapter son monitoring environnemental ?
- USP <922> Water Activity: A Better Approach for Lyo Moisture Determination. Applications enabled by rapid non-destructive headspace moisture analysis of freeze-dried product
- Gestion durable de l'eau dans l'industrie pharmaceutique
- Rapid Testing for Cell & Gene Therapy Products: A Three-Level Approach Using an Automated Solid Phase Cytometry System
What does 21 CFR Part 11 mean in everyday online analytics?
The article highlights some details how FDA 21 CFR Part 11 and EU Annex 11 shall be implemented and can be “lived” effectively. By reaching compliance more easily, operational efficiency is enhanced, and costs are lowered. You (The customer) is given peace-of-mind. The article gives answers to some key questions like “Can I fulfill 21 CFR Part 11 easily?” and “How do I reach data integrity?”

1. Introduction – FDA 21 CFR Part 11 and EU Annex 11
In pharmaceutical manufacturing, analytical data must be generated throughout the production cycle and stored for many years as evidence that pharmaceuticals are safe for consumption. These data can be archived as:
• paper records (hardcopies only)
• printouts linked to the underlying electronic records (hybrid)
• electronically signed electronic records (no paper).
There are many rules that must be followed for valid records. The requirements for the control of electronic records and signatures can be found in Title 21 CFR Part 11, issued by the U.S. Food and Drug Administration (FDA).
FDA 21 CFR Part 11 was created to enforce the correct and harmonized usage of electronic records and electronic signatures. As a result, electronic signatures were given legal equivalence with traditional “wet ink” signatures on paper. FDA 21 CFR Part 11 – sometimes just called Part 11 or 21 CFR et al. – is one of the most important regulations relevant to pharmaceutical manufacturing. The rules apply to all pharmaceutical products manufactured in the United States, and to products manufactured elsewhere but distributed in the United States, which gives it international relevance.

Very similarly, the European Union’s (EU) Annex 11 for computerized systems impacts manufacturers who export to the EU and those who manufacture products in the EU. Scrutiny of the parallel FDA and EU rules shows the authorities share a mutual intent to have safe, validated computer systems and qualified networks for drug and device manufacturing.
Compliance with FDA 21 CFR Part 11 and EU Annex 11 holds numerous advantages:
• Improved data integrity,
• Lower risk of regulatory violations,
• Simplified recording management,
• Increased operating efficiency.
The only short-term disadvantage of working with electronic records and following 21 CFR Part 11 – for companies coming from paper records – is the large effort to acquire new tools and set up new processes and having to implement stringent (electronic) user management and validate processes. In most cases the investment pays off.
In the following chapters only FDA 21 CFR Part 11 is referred to because it is more common and also more concrete for the implementation than EU Annex 11.
2. What can you expect from your instrument manufacturer?
A paperless world with fully electronic data handling promises cost savings from improved efficiency and reduced physical handling and storage. Paperless processes look appealing, make sense and are increasing in number. Most of the implementation of FDA 21 CFR Part 11 and EU Annex 11 lies with the end-user and must be defined and lived according to the company’s data governance rules.
While the operator has the full legal responsibility when developing and producing pharmaceutical products in accordance with an array of laws, rules and standards, instrument manufacturers can support the end-user with thorough implementation of these three features:
• user management on the instrument incl. electronic signatures.
• audit trail of all manipulations executed on the instrument.
• consistent and complete data incl. calibration history, meta data etc.
3. User management is key
Access can be controlled from the system level down to the object level, for example a single valve or a range of cleaning equipment or an entire ingredient list. Access to functional inputs can be limited, including the right to open a single valve, or start a CIP (Clean-in-Place) process, or schedule the next batches and campaigns.
Operators must identify themselves both at login and before an input is accepted; for example, before a motor is switched on or a cleaning process is started.
What does this mean for measuring and monitoring devices?
The pharmaceutical industry and related, similarly regulated industries, pursue solutions that protect data against unauthorized manipulation, may this manipulation be with good or bad intention, or unintentional.
It is important to realize that no system protects goods or intellectual property including information entirely, guaranteeing a level of 100% data security. Mistakes happen and industrial sabotage is reality. The best protection is given by “living” a company culture that propagates good data governance. Who is authorized to do what on your instruments? (Illustration 1)
Standard procedures to limit physical access to the monitoring equipment or related data lie in the responsibility of the pharmaceutical company. To protect your instrument from unqualified operation you can lock it up in a room where only the administrator has access, or you follow a user management as recommended by International Society of Pharmaceutical Engineering (ISPE) in their Good Automated Manufacturing Practice (GAMP) Guide 5, which means you implement:
• hierarchy levels,
• password complexity and history,
• username rules related to uniqueness and expiration.
For login, authentication, and electronic signing, two component security codes must apply. Every combination of user identification and password shall be unique.
The algorithms that check the minimum password length and complexity, password aging, and re-use of recent passwords must be secure and possibly configurable.

With regards to user hierarchy, there are simple solutions like the rule that Swan Analytical Instruments pursues:
1. Level 1 – administrator – can read, write and delete data, and create profiles.
2. Level 2 – maintenance manager – can read and write data.
3. Level 3 – operator – can merely read data.
4. Easy ways to achieve data integrity
Traceability means that an organization can trace who did what, when and possibly why? Data integrity goes a step further: if your organization follows the ALCOA, ALCOA+ or even ALCOA++ rules (see illustration 3) you have the guarantee that the data are complete, legible and that you are working with the original set of data. More importantly, this level of data quality is what the FDA auditors are expecting, and you will not get a warning letter. (Illustration 3)
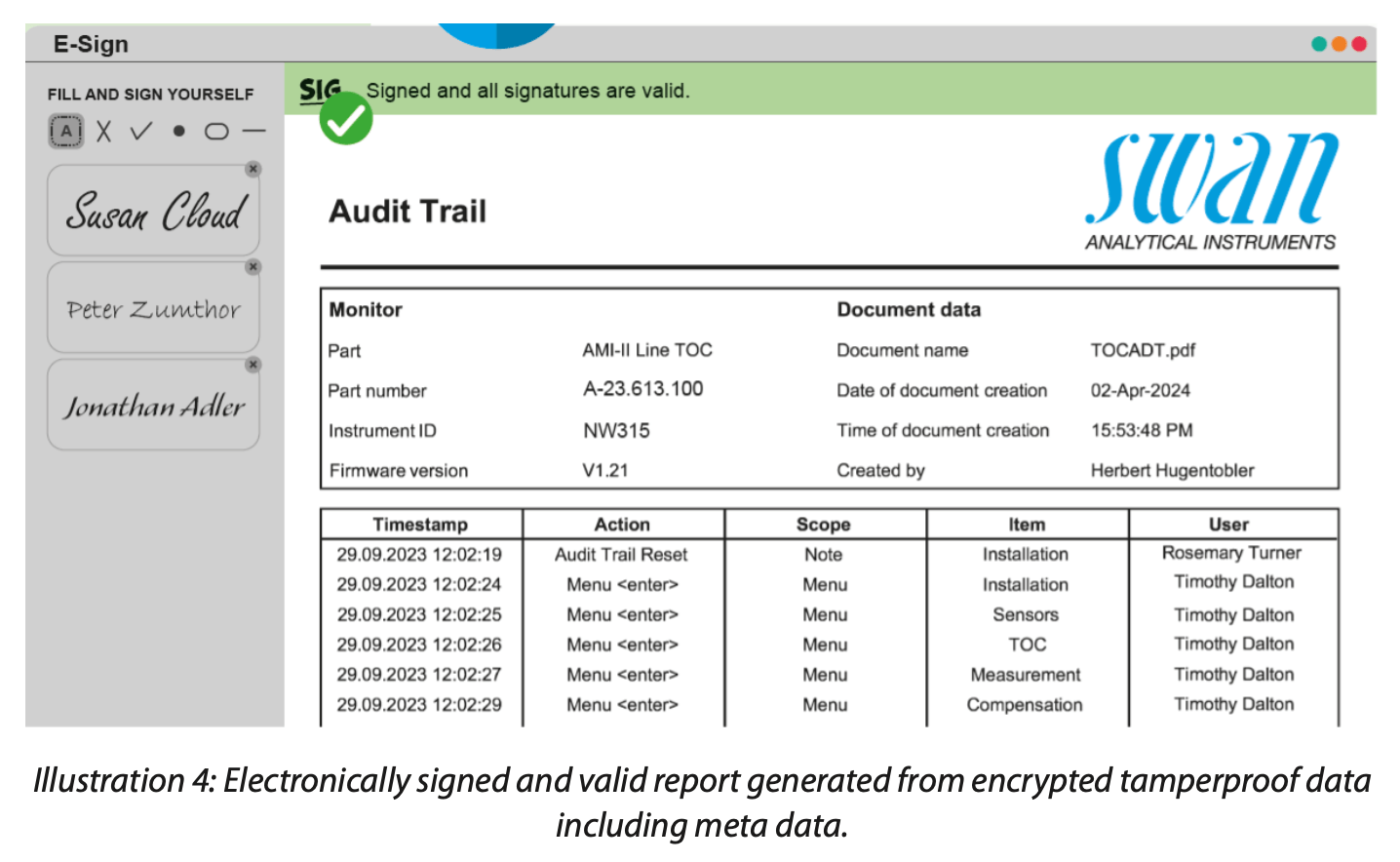
Solutions like the new Swan Guard PC software encrypt critical data, namely the audit trail and the calibration and SST (System Suitability Test) history, to make them secure. This tamperproof solution covers all meta data like time and date stamp, raw data, instrument ID and the action initiators’ names, following ALCOA++. Yet it is easy to use. What makes Swan Guard secure and, in many ways, unique are these features:
• The instrument generates encrypted data that can only be converted into certified, protected PDF reports.
• Electronic signing avoids unauthorized reporting (see illustration 4 as an example of a valid report).
• Any tampering of data, even if it is only one character, will result in a rejection of the report.
5. The right tools in place
Compliance always lies in the responsibility of the owner. A good analogy is your private car: The state requests you to pass street worthiness tests and the police might pull you over if they doubt your car is fit for purpose. You can take your car to the garage to have it checked and serviced but you must remember to do so! In addition, to keep your car in a good condition you must use it for the intended purpose, react to alerts and regularly check the air pressure in all four tires. You should also be aware who uses your car besides you. Finally, you must keep all legal papers in a safe place, valid and legible.
The same applies to measuring devices in pharmaceutical manufacturing. Clear roles and responsibilities and the respective processes must define how and by whom they are used and how they are maintained. With the right training of staff and necessary care in place the measuring device will yield the data quality that you need to produce your goods at a defined quality level while maximizing productivity. The level of data governance that your company pursues will determine whether your reach a compliant yet efficient process or not.
Partager l’article


Références
1. EMA: The European Medicines Agency (EMA) is a decentralized agency of the European Union (EU). It is responsible for the scientific evaluation, supervision and safety monitoring of medicines.
2. ISPE: The International Society for Pharmaceutical Engineering (ISPE) is a nonprofit association serving its members by leading scientific, technical, and regulatory advancement throughout the entire pharmaceutical lifecycle.
3. GAMP5, 2nd edition, The Good Automated Manufacturing Practice (GAMP) Guide for Validation of Automated Systems in Pharmaceutical Manufacture. The second edition of GAMP5 was released in July 2022.
4. EUDRALEX Rules: Governing Medicinal Products in the European Union, Volume 4, Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use.
