


Octobre 2024
La Vague n°83
EU GMP Annex 1 / Maitrise du risque patient ICH Q9(R1) / Digitalisation
Sommaire
- What does 21 CFR Part 11 mean in everyday online analytics?
- Digitalization in cleaning validation an overview of possibilities, challenges, and opportunities for savings?
- EU GMP Annexe 1. Mise en œuvre de la Stratégie de Contrôle de la Contamination
- Impact of the new Annex 1 on Sterile Filling
- The Challenges of Floor Cleaning & Sanitization
- Réduction énergétique des centrales de traitement d’air : comment adapter son monitoring environnemental ?
- USP <922> Water Activity: A Better Approach for Lyo Moisture Determination. Applications enabled by rapid non-destructive headspace moisture analysis of freeze-dried product
- Gestion durable de l'eau dans l'industrie pharmaceutique
- Rapid Testing for Cell & Gene Therapy Products: A Three-Level Approach Using an Automated Solid Phase Cytometry System
EU GMP Annexe 1. Mise en œuvre de la Stratégie de Contrôle de la Contamination
Depuis la parution de la mise à jour de l’Annexe 1 des Good Manufacturing Practices (GMP) ou Bonnes Pratiques de Fabrication (BPF) en 2022, la Contamination Control Strategy (CCS) ou stratégie de contrôle de la contamination (SCC) est au centre de l’attention. Cet article fait un rappel de sa structure, détaille l’utilisation de certains guides et l’utilisation par retour d’expérience pour mettre en place une CCS claire et efficace et supporter une bonne maitrise de la contamination.

Dans l’univers exigeant de l’industrie pharmaceutique, la garantie de la qualité, la sécurité et l’efficacité des médicaments est primordiale. Un aspect incontournable de cette garantie est la gestion de la contamination. Dans la mise à jour de 2015, les Good Manufacturing Practices (GMP) européennes ou Bonnes Pratiques de Fabrication (BPF) énoncent dans le chapitre 3 “Locaux et équipements” et le chapitre 5 “Production” les exigences réglementaires imposées pour minimiser les risques de contaminations. En 2023 la publication de la version révisée de l’Annexe 1 des GMP Européennes, qui traite des GMP pour les médicaments stériles, renforce encore ces exigences en mettant l’accent sur la prévention de la contamination.
Quand nous parlons de contamination, nous sous-entendons souvent contamination microbienne. Cependant, cette contamination dans l’industrie pharmaceutique peut prendre de nombreuses formes, y compris microbienne, particulaire et chimique, et peut provenir de diverses sources telles que les matières premières, les équipements, les processus de fabrication eux-mêmes et le personnel. Les conséquences d’une contamination non maîtrisée peuvent être importantes, allant de la perte de lots de production coûteux à des risques pour la santé des patients, y compris les conséquences réglementaires pour les fabricants. Face à ces enjeux, la fabrication de produits pharmaceutiques, en particulier la fabrication de produits stériles, requiert une grande attention pour éviter toute forme de contamination. C’est dans ce contexte que l’Annexe 1 a été revue et détaille les attentes réglementaires pour la fabrication aseptique, soulignant l’importance d’une Control Contamination Strategy (CCS) ou Stratégie de Contrôle de la Contamination (SCC), robuste et bien conçue.
La CCS est un document global expliquant les moyens mis en place pour démontrer que les risques de contamination sont sous contrôle et permettent de prévenir, détecter et éliminer les risques de contamination à toutes les étapes de la production pharmaceutique pour garantir la qualité des produits.
Elle englobe divers aspects, allant de la conception des installations et de l’équipement à la formation du personnel, en passant par les procédures opérationnelles et la surveillance environnementale. Pour les fabricants, se conformer à l’Annexe 1 n’est pas seulement une question de respect des lois ; c’est une démarche essentielle pour assurer la qualité des produits et protéger la santé des patients.
Dans cet article, nous allons aborder comment rédiger cette CCS. Nous commencerons par étudier brièvement le sujet de la contamination pour comprendre les attentes de la CCS. Nous détaillerons ensuite différentes approches proposées par les organisations ECA Foundation et Parenteral Drug Association (PDA ; en français : Association des médicaments parentéraux) ainsi que l’approche suivant les 5M et ferons une synthèse des pratiques à utiliser. L’objectif est la mise en œuvre d’une CCS efficace, en guidant les professionnels à travers le processus d’écriture et de mise en application de cette stratégie.
1. Comprendre la contamination dans l’industrie pharmaceutique
La contamination est l’introduction d’agents étrangers ou de substances dans un produit pharmaceutique, ce qui peut affecter sa qualité, sa sécurité et son efficacité. Dans l’industrie pharmaceutique, la contamination est classée en plusieurs catégories, microbienne, particulaire et chimique, chacune présentant des points d’attention pour la production et la qualité du produit.
La contamination microbienne est amenée par des bactéries, virus, levures ou moisissures. Ces micro-organismes peuvent être introduits dans le produit pharmaceutique à partir de l’environnement, des matières premières, de l’équipement ou du personnel. Cette contamination microbienne est particulièrement préoccupante pour les produits stériles, où la présence de tous microbes peut avoir des conséquences pour le patient.
La contamination particulaire est provoquée par la présence de particules étrangères inorganiques ou organiques, telles que poussière, fibres, particules métalliques ou reste de produits. Ces particules peuvent provenir de l’usure des équipements, de la manipulation des matériaux ou de la dégradation des composants du produit lui-même.
La contamination chimique ou contamination croisée se produit lorsque des substances chimiques ou biologiques d’un produit sont transférées à un autre, potentiellement en raison de pratiques de nettoyage inadéquates ou d’une mauvaise séparation des lignes de production.
Les sources potentielles de toutes ces contaminations seront étudiées et suivi dans le cadre de cette CCS. L’environnement de production avec l’air, les systèmes de renouvellement d’air, l’eau et ses systèmes de traitement, les surfaces et l’état général de propreté des installations dans les zones de production peuvent être des vecteurs de contamination. De même, l’état général des équipements et machines, leur état de nettoyage, les résidus de production précédente, l’usure des machines et le manque d’entretien peuvent également contribuer à la contamination. Quant aux matières premières utilisées pour fabriquer des produits pharmaceutiques, elles peuvent être contaminées à la source ou pendant le stockage et le transport. Enfin le personnel lui- même peut introduire des contaminants par contact direct ou indirect avec le produit ou les surfaces de contact du produit.
Le nombre de sources potentielles de contamination est longue mais chacune est importante et doit être considérée et contrôlée. Chaque manquement peut entrainer une contamination avec des conséquences, allant de l’inefficacité thérapeutique et des effets secondaires à des incidents de sécurité des patients, tels que des infections ou des réactions toxiques.
2. La CCS dans l’Annexe 1
Cela souligne l’importance d’une CCS pour préserver toutes les étapes de productions pharmaceutiques contre les risques de contamination. La définition de la CCS selon l’Annexe 1 est “Un ensemble de moyens de maitrise prévus pour les microorganismes, endotoxines/pyrogènes et particules, issus de la connaissance actuelle des produits et des procédés, qui assure la performance des procédés et la qualité du produit. Les moyens peuvent comprendre des paramètres et des attributs liés aux matières premières, aux composants et produits stériles et aux conditions de fonctionnement des installations et équipements, aux contrôles en cours de fabrication, aux spécifications des produits finis, ainsi qu’aux méthodes associées et à la fréquence de surveillance et de contrôle”.
L’Annexe 1 renseigne sur le contenu de la CCS. Elle liste les sujets à aborder qui reprennent l’ensemble des éventuelles sources de contamination à analyser dans ce document support. Elle ne dit cependant pas comment construire et structurer ce document dans le cas d’une nouvelle installation à mettre en place ou pour une installation en opération avec de la documentation déjà existante.
Les 16 points repris dans l’Annexe 1 couvrent les concepts suivants :
1. Conception de l’installation et du processus
2. Locaux et équipement
3. Personnel
4. Utilités
5. Contrôle des matières premières – IPC
6. Conteneurs et fermetures des produits
7. Qualification des fournisseurs
8. Gestion de activités sous-traitées
9. Gestion des risques de processus
10. Validation des processus
11. Validation des processus de stérilisation
12. Maintenance préventive
13. Nettoyage et désinfection
14. Systèmes de surveillance
15. Prevention – analyse de tendances, investigation et CAPA 16. Amélioration continue.
La rédaction d’une CCS est un exercice qui peut être compliqué pour les industriels et plusieurs raisons peuvent se combiner pour transformer l’exercice en défi difficile à relever. En effet, les processus de fabrication des produits pharmaceutiques peuvent être extrêmement complexes, impliquant de nombreux équipements, technologies et étapes. Identifier et évaluer les risques de contamination à chaque étape, nécessite une expertise technique et une compréhension des interactions entre ces différents composants du processus de fabrication. La validation des mesures de contrôles de la contamination, peut ainsi être un processus long et difficile pour démontrer sa nécessité et être accepté par chaque service impliqué.
Pour les nouvelles installations ou les entreprises qui lancent de nouveaux produits, il peut y avoir un manque de données historiques sur lesquelles s’appuyer pour évaluer les risques de contamination. De plus, la mise en place d’une CCS efficace peut nécessiter des ressources significatives en termes de temps, de personnel et de finances. Coordonner ces efforts à travers différents départements (par exemple, production, assurance qualité, ingénierie, etc.) et obtenir l’engagement de toutes les parties prenantes peut être difficile tout en assurant la continuité des activités quotidiennes.
Plusieurs approches ont été proposées aux industriels pharmaceutiques par des associations accompagnants les entreprises pour aider à l’élaboration d’une CCS et pour les accompagner dans la mise en place et l’écriture de ce document.
3. La rédaction de la CCS selon ECA Foundation
ECA Foundation, une organisation à but non lucratif accompagnant l’industrie pharmaceutique à l’échelle européenne, propose une aide pour le développement d’une CCS. A travers la ECA Task Force on Contamination Control Strategy ( Task force de la ECA sur la stratégie de contrôle de la contamination), elle a publié en 2022 un document pour aider les responsables respectifs à élaborer une CCS :“How to Develop and Document a Contamination Control Strategy” (“Comment développer et documenter une stratégie de contrôle de la contamination”) ?
Ce document fournit une description sur la façon d’élaborer et documenter une stratégie efficace. Il détaille une approche en trois phases : le développement ou revue de la CCS, la compilation des documents concernant la CCS et enfin son évaluation. Ces trois phases reprennent la même approche que les trois phases de la validation des processus énoncées par la FDA.
Pour l’élaboration de la CCS, cette organisation identifie une approche soit pour une nouvelle installation soit pour une installation existante en fonction du degré de compréhension des processus. Pour une nouvelle installation, il sera nécessaire de commencer par s’approprier le processus, le comprendre et le cartographier pour identifier les sources potentielles de contamination. L’évaluation de ces sources potentielles via une analyse de risque permettra de classifier les dangers de contamination possibles et d‘identifier les mesures préventives à mettre en place. Ces moyens de contrôle cités dans la CCS devront être décrits dans la documentation. Pour une installation existante ou bien avec une connaissance approfondie du processus, la CCS regroupera et fera la synthèse des mesures de contrôle déjà existantes et analysera les éventuels écarts. L’analyse de risque sera une mise à jour ou bien un complément par rapport aux documentations déjà en vigueur et permettra d’identifier les mesures supplémentaires à mettre en place au besoin.
Cette organisation décrit une méthode de travail reprenant en trois étapes ce qui doit être fait sur chaque sujet impactant le contrôle de la contamination tel que défini dans l’Annexe 1.
La phase 1 décrit l’ensemble de la documentation, analyse de risque, rationnel, mesure de contrôle en place pouvant être écrite ou utilisée pour garantir le contrôle de la contamination. L’ensemble de ces analyses se fait par rapport à la liste des seize éléments devant être repris dans la CCS comme décrit dans l’Annexe 1. Des liens sont repris avec l’Annexe 1 pour expliquer et mettre en avant les points d’attention sur des pratiques de contrôle à mettre en place.
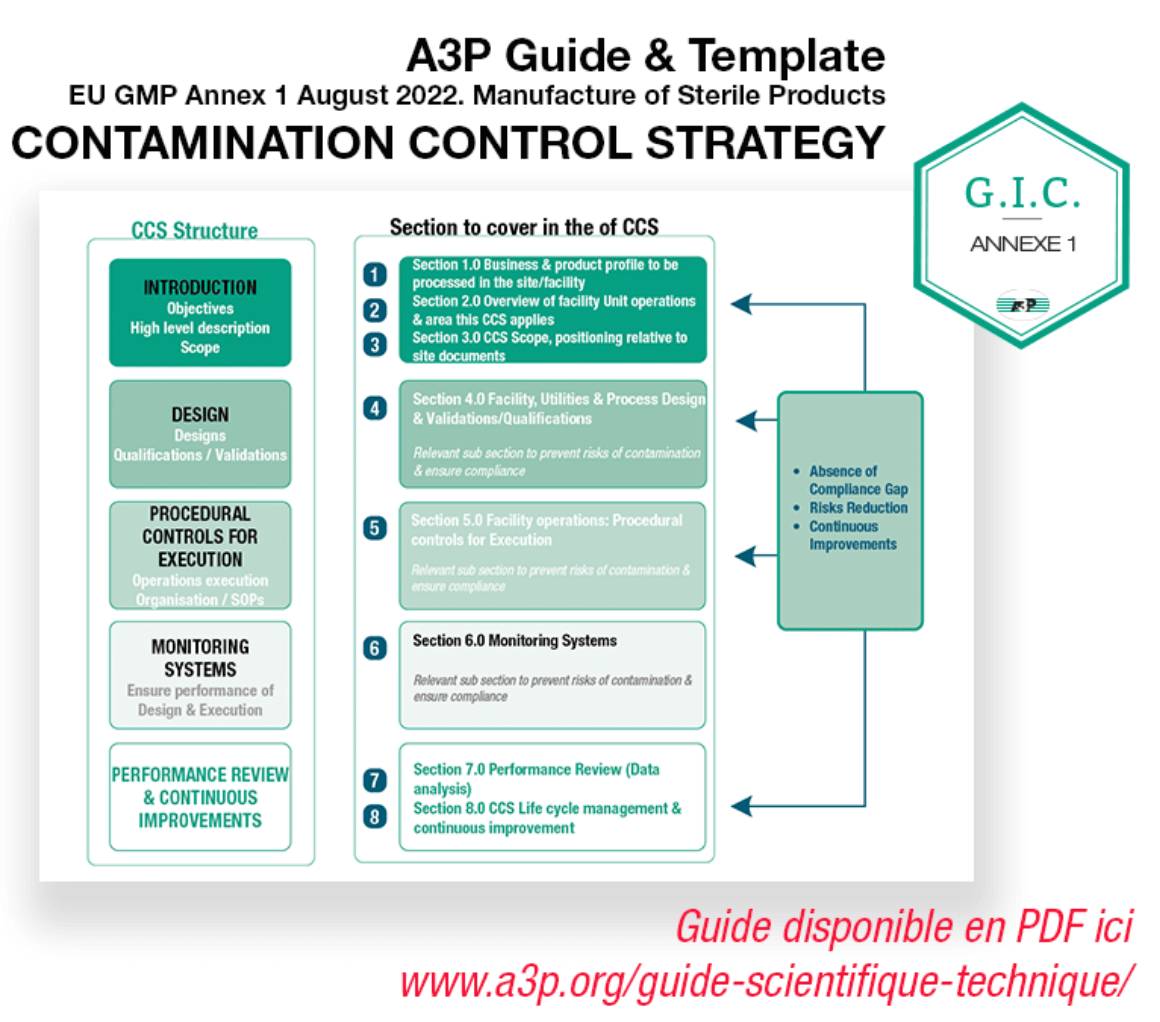
La phase 2 décrit comment compiler tous ces documents en un document CCS unique reprenant de façon lisible et compréhensible l’ensemble de la documentation identifiée précédemment. Un sommaire et une trame de CCS sont détaillés dans les annexes de ce document.
La phase 3 décrit le cycle et les conditions de revu de ce document pour le conserver à jour et l’aligner avec le niveau de contrôle approprié pour garantir le contrôle de la contamination de l’installation.
L’avantage de ce document support pour réaliser une CCS est de bien mettre en avant l’ensemble de la documentation existante et les analyses de risques pour justifier l’ensemble des moyens de contrôle et donc la documentation regroupée pour expliquer le contrôle de la contamination.
4. La rédaction de la CCS selon la PDA
L’association PDA, fournissant des guides pour faciliter l’amélioration du développement et la fabrication de produits pharmaceutiques parentéraux, a édité le rapport technique PDA n° 90 : “Développement d’une stratégie de contrôle de la contamination” (TR-90) en février 2023. Il y fournit des conseils sur la façon d’établir une CCS efficace et décrit une gouvernance à mettre en place avec trois niveaux interdépendants inclus par le système de la qualité pour le succès d’une bonne maitrise de la contamination.
Le premier niveau incluant les éléments individuels décrit les attentes pour minimiser les risques de contamination. Cela inclut les éléments fondamentaux tels que la conception et la construction des installations de production, la conception des flux d’air, la sélection des matériaux de construction, la disposition des équipements, le choix des matières, consommables et fournisseurs, les méthodes de nettoyage ou de stérilisation et les formations définies pour le personnel.
Le second niveau incluant les processus qualité mis en place de qualification et validation de ces éléments individuels permet de démontrer que chaque élément individuel sélectionné peut raisonnablement atteindre le niveau de contrôle approprié. L’objectif est de créer un environnement contrôlé qui empêche l’introduction, la génération et la rétention de contaminants.
Le troisième niveau comprend les surveillances mises en place telles que la surveillance de l’air, des surfaces, de l’eau, du personnel et d’autres paramètres critiques pour détecter rapidement toute déviation par rapport aux normes établies. Les surveillances mises en place peuvent être par exemple les analyses de tendances, ou des alarmes. Les données recueillies sont utilisées pour évaluer l’efficacité des contrôles environnementaux et pour prendre des mesures correctives si nécessaire. Une trame est proposée en annexe du rapport technique pour aider à la rédaction de cette CCS.
L’avantage de ce document est sa structure qui permet d’organiser chaque partie de façon individuelle et de la suivre à travers les différents niveaux.
Il faut démontrer pour chaque élément individuel, quelle est la donnée critique identifiée et démontrer qu’elle est bien représentative d’un risque de contamination. Il faut ensuite la suivre dans le temps pour garder sous maitrise cet élément. Pour défendre l’efficacité du document, chaque élément individuel doit être considéré de manière holistique car ils sont interdépendants les uns des autres. Par exemple, une conception d’installation bien pensée peut être compromise par de mauvaises pratiques opérationnelles, et même les meilleures pratiques opérationnelles ne peuvent pas compenser une conception d’installation inadéquate.
5. La rédaction de la CCS en suivant les 5M
L’écriture d’une CCS peut aussi se structurer autour d’une analyse de risque des sources de contamination menée selon un diagramme 5M (ou diagramme d’Ishikawa). Le diagramme reprenant les 5M permet d’identifier les sources potentielles de risque de contamination en fonction de chaque catégorie : Matière première, Matériel, Main d’œuvre, Milieu, Méthode.
La branche Matière première permettra d’identifier les risques de contamination en fonction de la qualité de la matière et du fournisseur, son stockage et le flux d’entrée des matières en zone de production jusqu’à leur utilisation.
La partie Matériel correspondant aux équipements utilisés dans la production de façon directe ou indirecte, permettra d’identifier des risques de contamination issus d’activités tels que le nettoyage, la maintenance mais aussi leur assemblage dans des connections aseptiques.
La branche Main d’œuvre permettra d’identifier les risques de contamination provenant du personnel incluant, par exemple, l’hygiène, la formation, la traçabilité du personnel intervenant en zone aseptique, et les séquences d’habillage.
La branche Milieu correspondant aux locaux en lien avec la production permettra d’identifier les risques de contamination issus, par exemple, du nettoyage et désinfection de ces locaux incluant les zones blanches de tous grades D à A, ou du système d’arrivée d’air contrôlé.
Enfin, la branche Méthode permettra d’identifier les risques de contamination au cours du processus de production lui-même incluant par exemple les mélanges, les filtrations, et les filtrations finales.
La cotation de chacun de ces risques identifiés par branche 5M permettra de définir les risques critiques. Basés sur cette cotation et avec justification, des moyens de contrôle seront établis pour mettre sous contrôle, diminuer ou éliminer les risques.
L’avantage principal de cette méthode est qu’elle offre une approche structurée pour examiner les différentes dimensions qui peuvent influencer la qualité d’un produit ou la performance d’un processus.
L’analyse branche par branche encourage l’examen de tous les aspects du processus de production, ce qui permet d’identifier des causes de problèmes qui pourraient être négligées. Elle est aussi relativement simple à comprendre et à mettre en œuvre, ce qui la rend accessible à tous les niveaux de l’organisation.

6. La structure de la CCS
L’objectif de la CCS est de documenter dans une approche globale les mesures de contrôle organisationnelles, techniques et procédurales mises en place et de garantir que tous les risques de contamination de micro-organismes, endotoxines/pyrogènes et particules sont identifiés, évalués et atténués de manière appropriée.
L’objectif de la rédaction de cette CCS est de pouvoir répondre au “pourquoi“, c’est-à-dire de comprendre non seulement comment la contamination se produit, mais surtout pourquoi elle se produit dans un processus, et par conséquent dans le produit. Pourquoi une contamination peut se produire, pourquoi à cet endroit, pourquoi peut-elle proliférer, dans l’équipement, le process, l’installation, etc.
Répondre à l’ensemble de ces “pourquoi” permettra de comprendre les causes racines et donc de définir l’outil de mitigation optimal pour diminuer voire éliminer le risque de contamination, et de mettre en place des solutions durables.
Cette CCS peut se structurer, soit d’un document principal global incluant tous les aspects de la stratégie de contrôle mis en place, soit d’un document chapeau se référant à des documents connexes distincts répartis sur l’ensemble des matières à aborder et devant être reliés ensemble. Elle ne doit pas être une simple liste des documents existants dans l’installation ou une liste de documents reprenant les points cités dans l’Annexe 1. C’est un document global, qui met en avant le risque et la justification des contrôles mis en place en vue de démontrer que tous les dangers de contamination sont maitrisés ; il permet de relier les différents aspects et les mesures d’accompagnement associés.
Quelle que soit l’approche sélectionnée, quel que soit l’historique de l’installation, la première étape dans l’élaboration d’une CCS sera l’évaluation des risques. Cela implique l’analyse systématique de tous les processus de production pour identifier, classer et gérer les risques potentiels de contamination en fonction de la probabilité et la sévérité des différents scénarios de contamination. Il sera nécessaire de bien définir le ou les processus ou l’installation concernées ainsi que les produits stériles et/ou tout produit ou ingrédient nécessitant un contrôle de la charge biologique. Il ne faudra pas confondre l’analyse de risque de ou des processus avec l’analyse de risque des sources de contamination ; la première permettant d’identifier les risques potentiels pouvant avoir un impact sur la qualité du produit et la seconde permettant d’identifier les sources des contaminations potentielles de l’installation. Cette activité devra être réalisée avec une équipe multidisciplinaire avec des membres de différents départements tels que la production, l’assurance qualité, et l’ingénierie. Les analyses de risques réalisées ou identifiées si existantes, les mesures préventives seront ensuite mises en place pour contrôler et surveiller ces points critiques identifiés. Cette démarche permet de garantir que les mesures de contrôle mises en place sont à la fois adéquates et proportionnées à la nature et à la gravité des risques identifiés. Sur la base de cette ou ces analyses des risques, la CCS doit décrire cette stratégie de manière claire et détaillée. Elle reprendra comment chaque risque sera géré ou atténué et détaillera la justification des choix faits, des descriptions des contrôles mis en place, et des procédures pour la surveillance et la révision de la stratégie. Cela peut inclure des changements de conception, des Standard Operating Procedures (SOP) ou procédures opérationnelles standard, des programmes de formation, des pratiques de nettoyage améliorées, et des systèmes de surveillance environnementale.
La stratégie mise en place doit permettre une approche proactive, permettant de détecter toute tendance ou toute anomalie en amont d’évènement et où la prévention est priorisée par rapport à la réaction face aux incidents de contamination.
7. Conclusion
Comme nous l’avons exploré dans cet article, une CCS efficace repose sur une évaluation minutieuse des risques, une conception intelligente des installations, un contrôle environnemental rigoureux, des procédures opérationnelles standardisées et une formation approfondie du personnel. En suivant les étapes décrites pour la mise en place de cette stratégie et la rédaction de ce document, la CCS correspondra aux attentes qualité et support par rapport à la gestion de risque de contamination pour maintenir une qualité constante des produits pharmaceutiques. Elle garantit que les points critiques de contrôle de la contamination sont identifiés et que les mesures de contrôle de la contamination sont intégrées dans le processus de fabrication. De plus, elle permettra d’identifier et de gérer proactivement les risques de contamination, ce qui peut réduire la fréquence des incidents de contamination, les rappels de produits, et les pertes financières associées.
Alors que l’industrie pharmaceutique continue d’évoluer et de relever de nouveaux défis, la CCS devient un document incontournable de la qualité pharmaceutique. Les entreprises qui investissent dans des stratégies de contrôle de la contamination robustes et qui adoptent les dernières innovations sont mieux équipées pour protéger leurs patients, leurs produits et leur réputation. La mise en œuvre de la CCS selon l’Annexe 1 fournit ce cadre exhaustif pour garantir la production de médicaments stériles sûrs et efficaces.
Partager l’article


Références
1. EU GMP Annex 1, “Manufacture of Sterile Medicinal Products”, August 2022
2. ECA Foundation Guidance Document, “How to Develop and Document a Contamination Control Strategy”, January 2022
3. PDA, Parenteral Drug Association, Technical Report “TR No.90 CCS Development in Pharmaceutical Manufacturing”, 2023
