


Sommaire
- Les enjeux industriels de la généralisation de l’adoption des systèmes à usage unique (SUS : Single-Use Systems).
- Current state & trends for Single-Use technologies implementation in the Biopharmaceutical Industry.
- Continuous Processing. Performance Enhancements for Perfusion Applications in 50L to 500L Single-Use Bioreactors: A Technical Comparison of Performance Characterization, Cell Culture & Scale-Up Modeling.
- Implementation of Single-Use in Drug Substance filling before transportation: Product Development case study.
- Technologies de connexion à usage unique : situation actuelle et tendances.
- Extractables and Leachables from SUS - aspects beyond Extractables Measurement & standardization.
- Evaluation toxicologique des extractibles et relargables liés à l'utilisation des Systèmes à Usage Unique (SUS).
Les instances réglementaires attendent des sociétés pharmaceutiques qu’elles garantissent l’efficacité et l’innocuité des médicaments qui sont dispensés aux patients. Depuis que l’utilisation des Systèmes à Usage Unique, appelés également “disposables”, dans les procédés de fabrication devient une pratique courante, les industriels doivent apporter la preuve qu’aucune substance toxique issue des matériaux constitutifs de ces dispositifs ne migre dans le produit fini.

La multiplicité des matériels, leur composition et les conditions d’utilisation rendent difficile l’évaluation. C’est pourquoi une démarche structurée d’évaluation par le risque permet de prioriser les études à mener pour en sécuriser l’usage.
Dans le cadre du dépôt du dossier d’autorisation de mise sur le marché, les études d’interactions entre le conditionnement primaire et le médicament sont requises de manière à apporter la preuve que des relargables potentiellement toxiques issus du contenant, quelle que soit sa nature, ne seront pas administrés au patient. Contrairement aux contenants en verre qui sont, dans la plupart des cas, relativement “neutres” par rapport aux produits, les composants en plastique (seringues, flacons, blisters) ou en élastomère (bouchons, joints de pistons), en contact direct avec les produits, sont moins inertes et présentent plus de risque de relargage. Différentes lignes directrices réglementaires européennes(1) et américaines(2) définissent les informations nécessaires à produire pour la qualification des articles de conditionnement. En complément à ces textes, les pharmacopées spécifient les caractéristiques des différents polymères couramment utilisés ainsi que les tests à réaliser pour justifier de leur compatibilité avec les produits.
Certains composants polymériques utilisés lors du procédé de fabrication font également l’objet d’études systématiques de relargables lorsque qu’aucune étape postérieure ne permet l’élimination d’impuretés potentielles générées par la dégradation des différentes pièces constitutives du dispositif. C’est le cas, par exemple, des filtres utilisés pour la stérilisation finale de produits injectables dont la qualification doit apporter la preuve qu’ils ne sont pas source de contamination particulaire ou chimique.
Pour apporter plus de flexibilité dans la production et limiter les risques de contaminations croisées, certaines sociétés ont opté pour l’utilisation des systèmes à usage unique (SUS) pour la fabrication et le stockage des produits ou des intermédiaires à différents stades du procédé. Le remplacement d’équipements en inox ou en verre par des matériels principalement constitués de polymères n’est pas sans impact sur la sureté du produit fini. En effet, les conditions dans lesquelles ces composants sont utilisés (température, pression, ph, …) peuvent entrainer une dégradation accélérée des matériaux constitutifs et apporter des impuretés éventuellement toxiques qui ne seraient pas forcément éliminées lors des étapes de purification.
La diversité des polymères, des additifs (antioxydants, plastifiants, catalyseurs) et des solvants entrant dans la composition des disposables ainsi que l’accumulation des impuretés potentielles tout au long du processus sont des facteurs qui doivent être pris en compte pour évaluer le risque associé à leur présence dans le médicament. La multiplicité des sources potentielles de contaminants rend difficile l’identification et la quantification des impuretés relarguées par les dispositifs polymériques mis en œuvre pour la fabrication. En l’absence de textes réglementaires spécifiques à l’usage des SUS, différentes associations se sont créées pour aborder la problématique liée aux extractibles et aux relargables (eg : PQRI, BPOG, PDA, BPSA, GIC A3P SUS, le magazine La Vague…) et apporter un support méthodologique et scientifique à l’évaluation des disposables.
En application du processus décrit dans l’ICH Q9(3), une évaluation des risques doit être menée pour chaque étape du procédé pour laquelle un SUS est utilisé. Cette estimation du risque va prendre en compte la sensibilité du matériau à l’extraction (type de polymère), la capacité d’extraction du produit (solide, liquide, nature et composition du contenu, …) et les conditions d’utilisation du composant (température, durée de contact, ratio volume/surface, pH…)(4 – 5 – 6). Le but de cette phase est clairement de catégoriser les disposables en fonction du réel risque de relargage de manière à adapter les actions de mises sous contrôle et en particulier à prioriser les études d’extractibles et relargables à mener. Le processus d’analyse doit être adapté au niveau de risque et doit permettre de statuer clairement sur la sécurité des systèmes utilisés. A titre d’exemple, les différentes phases d’évaluation pour les composants à risque moyen sont décrites dans le schéma.
Pour rappel, pour les composants à risque faible les études d’extractibles ne sont pas requises. Est attendue uniquement la conformité aux tests de la Pharmacopée Européenne et aux tests biologiques de l’USP(7 – 8) Classe VI.
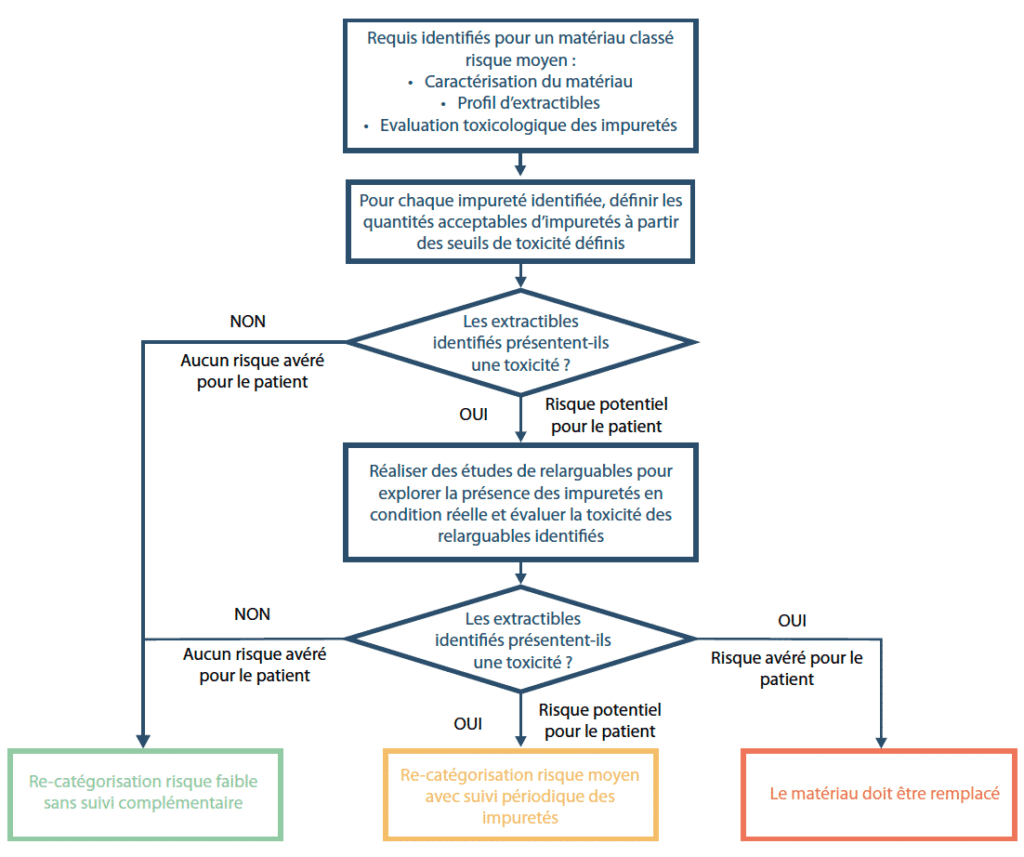
Pour les composants à risque moyen ou élevé, l’identification et la quantification des impuretés issues du polymère en contact avec le produit est nécessaire de manière à évaluer le risque associé à leur toxicité (9). Les informations sur les potentielles impuretés issues des composants peuvent provenir soit des dossiers de validation des fournisseurs soit d’études d’extractibles réalisées en conditions de simulation agressives pour le matériau. À titre d’exemple, le BioPhorum Operations Group (BPOG) propose une stratégie d’essais à réaliser pour l’étude des extractibles en spécifiant les solvants modèles à utiliser et les conditions opératoires à suivre en fonction de l’utilisation des SUS(10). Si des extractibles identifiés présentent une toxicité, des études de relargables doivent être menées pour éventuellement démontrer que cet extractible toxique n’est, en fait, pas présent dans le produit fini.
Parmi les volatiles (solvants, résidus de synthèse, monomères), semi-volatiles (additifs, produits de dégradation), non volatiles (principalement organiques, antioxydants, acides gras, stabilisants) et des impuretés élémentaires (métaux) identifiés, une première sélection de substances à risque peut et doit être faite, sur la base des données de sécurité, de manière à ne prendre en considération que celles qui présentent un risque pour le patient. Les critères de choix sont définis en fonction de leur nature et explicités dans les paragraphes ci-après.
Pour les impuretés élémentaires, l’ICH Q3D(11) stipule celles qui doivent être prises en compte lors de l’analyse (toxiques de Classe 1 et de Classe 2B), celles qui ont une toxicité avérée qui doivent être considérées si elles sont ajoutées intentionnellement (Classe 2B) ou celles qui présentent une toxicité en fonction de la voie d’administration (Classe 3). Les impuretés élémentaires de Classe 4, quant à elles, ne présentent aucune toxicité et dans ce cas seront exclues de l’analyse. Le guide, spécifie également les doses admissibles des éléments (PDE – Permitted Daily Exposure) qui permettent de calculer les concentrations limites acceptables dans le produit final.

Une impureté détectée dans le produit fini à un taux de 30% inférieur à la PDE est considérée négligeable et l’ICH Q3D(11) spécifie qu’elle ne nécessite pas de suivi particulier. Pour des taux allant de 30% à 90% de la PDE, compte tenu de la variabilité potentielle de la concentration en relargables, des contrôles périodiques réguliers sur des lots de produit fini peuvent être mis en place pour surveiller et confirmer le non dépassement des seuils acceptables. C’est seulement à l’issue de cette étape de surveillance et si les seuils ne sont jamais dépassés que le suivi régulier peut être abandonné et que le risque pourra être recatégorisé en risque faible. Si les dosages montrent des valeurs autour de la PDE ou supérieures, le risque potentiel pour le patient doit être réévalué au cas par cas. Si le risque est avéré, des moyens de réduction de l’impureté doivent être recherchés et, en leur absence, le matériau constitutif du “disposable” devra être remplacé.
Cette approche est également applicable pour les solvants résiduels pour lesquels l’ICH Q3C(R5)(12) les classe en fonction de leur toxicité et définit les PDE. La concentration limite en solvant est déterminée selon la formule :

Les règles d’évaluation des risques appliquées pour les impuretés élémentaires peuvent également s’appliquer pour les solvants résiduels.
Concernant les extractibles ou relargables qui ne sont pas couverts par les guides ICH, en fonction de leur concentration, une évaluation toxicologique est nécessaire pour statuer si ces composés présentent un risque réel pour le patient et définir les seuils acceptables dans le médicament. L’analyse toxicologique ne doit pas se limiter aux tests de toxicité aigüe, chronique et subchronique (DL50 relative à la voie d’administration du médicament), certes les indicateurs de la classe de la substance considérée, mais doivent également être intégrés des données issues de la littérature concernant le potentiel cancérogène, mutagène et toxique pour la reproduction. En fonction de la typologie du risque toxicologique identifié et du mode d’administration, deux seuils sont possibles. Pour les impuretés à potentiel cancérogène ou mutagène, la PQRI(13) a fixé la limite de toxicité (SCT – Safety Concern Threshold) à 0,15 µg/jour pour la voie nasale ou inhalée ainsi que pour les substances hautement cancérogènes et à 1,5 µg/jour pour les autres voies d’administration. Pour les substances ne présentant pas de risque oncogène ou mutagène avéré, le SCT a été défini indépendamment de la forme galénique, par défaut, à 1,5 µg/jour pour chaque impureté pour prendre en compte les composés pour lesquels aucune information toxicologique n’était disponible. Ces valeurs ont été établies par l’analyse de données scientifiques issues de bases de données toxicologiques comme la HSDB (Hazardous Substances Data Bank) et IRIS (Integrated Risk Information System). Compte tenu du facteur de sécurité appliqué au SCT, les relargables présents au-dessous de ce seuil ne nécessitent pas d’évaluation supplémentaire. Pour sélectionner les extractibles ou relargables à prendre en compte lors de l’analyse, un seuil analytique en µg/g de contenant (AET – Analytical Evaluation Threshold) est défini selon le calcul :

Dans le cas où le contenant est un assemblage de différentes pièces, c’est le poids de l’intégralité du dispositif qui doit être considéré pour le calcul de l’AET. Un facteur correctif peut être appliqué pour tenir compte de la variabilité de la méthode de traitement des échantillons et de l’incertitude de la méthode de contrôle. La détermination précoce de l’AET sur la base de données fournisseurs ou d’éléments bibliographiques est utile car l’identification et la quantification des impuretés majeures détectées lors des études d’extractibles et relargables ne sont pas requises au-dessous de ce seuil.
Les analyses de risques mises en œuvre pour catégoriser les risques permettent clairement d’identifier les composants qui nécessitent une évaluation pour sécuriser leur utilisation. Au vu de la difficulté d’obtention d’informations fiables ou représentatives de la part des fournisseurs sur leurs produits, l’analyse doit permettre de définir les études d’extractibles et relargables à mener pour garantir l’absence de contaminants chimiques toxiques issus des matériels en contact produit.
L’évaluation toxicologique des impuretés identifiées est une étape essentielle de l’analyse afin de caractériser le risque et de définir leurs seuils acceptables dans le médicament et permettre la mise en place d’une stratégie de surveillance adaptée.
Comme pour les articles de conditionnement primaire, il est clair que toute modification de la composition des polymères constitutifs des SUS ou de leur procédé de fabrication doit être géré dans le cadre de la maitrise des modifications et faire l’objet d’une revue du risque intégrant de nouvelles études et une analyse des données toxicologiques des substances détectées.
Partager l’article


Roland OLLIVIER – AKTEHOM
roland.ollivier@aktehom.com
References
(1) Guideline on plastic immediate packaging materials CPMP/QWP/4359/03 du 29 mai 2005
(2) Guidance for Industry et Code of federal register 21CFR – Container closure systems for packaging human drugs and biologics (1999)
(3) ICH Q9: Quality Risk Management. US Fed. Reg. 71(106) 2006
(4) Bennan J. et al, BioPharm International December 2002, 22-34; Evaluation of Extractables from Product-Contact Surfaces
(5) BPOG Best Practices Guide for Evaluating Leachables Risk in Biopharmaceutical Single-Use Systems
(6) The 2010 BPSA Recommendations for Testing and Evaluation of Extractables from Single-Use Process Equipment
(7) USP <87> Biological Reactivity, In Vitro
(8) USP <88> Biological Reactivity Tests, In Vivo
(9) Brochard T.H. et al, Regulatory Toxicology and Pharmacology; 2016 Nov; 81:201-211; Assessing safety of extractables from materials and leachables in pharmaceuticals and biologics – Current challenges and approaches
(10) Ding W. et al, Pharmaceutical engineering 34, 1 – 11, 2014; Standard extraction protocol for Single-Use systems in biopharmaceutical manufacturing
(11) ICH Q3D(R1) – Guideline for elemental impurities – 2018 (Step 2)
(12) ICH Q3C(R6) – Impurities: Guideline for residual solvents – 2016
(13) PQRI Safety Thresholds and Best Practices for Extractables and Leachables in orally Inhaled and Nasal Drug Products – September 2006
Glossary
AET: Analytical Evaluation Threshold
BPOG: BioPhorum Operations Group
BPSA: Bio-process Systems Alliance
HSDB: Hazardous Substances Data Bank
IRIS: Integrated Risk Information System
PDA: Parenteral Drug Association
PDE: Permitted Daily Exposure
PQRI: Product Quality Research Institute
SCT: Safety Concern Threshold
