


Summary
- Data Integrity: towards the introduction of Data Management
- The link between regulation, quality system & Data Integrity
- Migration in the Cloud & Data Integrity
- Implications of calculating the PDE as the exposure limit for the analysis of risks in shared installations
- The IVDR signals the overhaul of the in vitro diagnosis industry
- Tungsten in the production of prefillable syringes. Also possible without tungsten
The publication in March 2018 by the MHRA of its new guide on the subject is the opportunity to recall the main objective of Data Integrity: to have confidence in the quality and integrity of the data that is generated, and to be able to reconstruct the activities. This article offers an interpretation of the main requirements and clarification on their implementation.
The control of Data Integrity has become a major concern of the health authorities, and no fewer than 6 guidelines1 have been published since 2016 on the subject by different regulatory bodies. These guidelines, some of which are still in draft form, show some convergence in their expectations. The detection of numerous violations of good manufacturing practices in terms of data control and outright frauds in the past, led the inspectors to clarify their position on this issue, and to educate themselves in this area. The number of Warning Letters, Non Compliance Reports and other orders on this question has thus increased considerably in recent years.
Data Integrity
Data integrity is commonly defined as being the extent to which critical data remain complete, consistent and accurate, throughout their life cycle. Although this is not a recent concept – it was already defined by the IEEE in the 1990s using the same wording, and Warning Letters issued by the FDA in the early 2000s can be found using the same terms – nevertheless its control requires new provisions, as a result of the growing digitization of processes, globalization of activities, and the multiplication of computerized systems through which data pass.
Reliable Decision-Making
For a decision to be robust, the integrity of the data that support it must be guaranteed. What confidence can be placed in a pharmaceutical decision taken on the basis of erroneous data?
It is then essential that an organization’s quality system can allow identification and control of the weak points of critical data, whether these data are electronic or recorded as a hard copy.
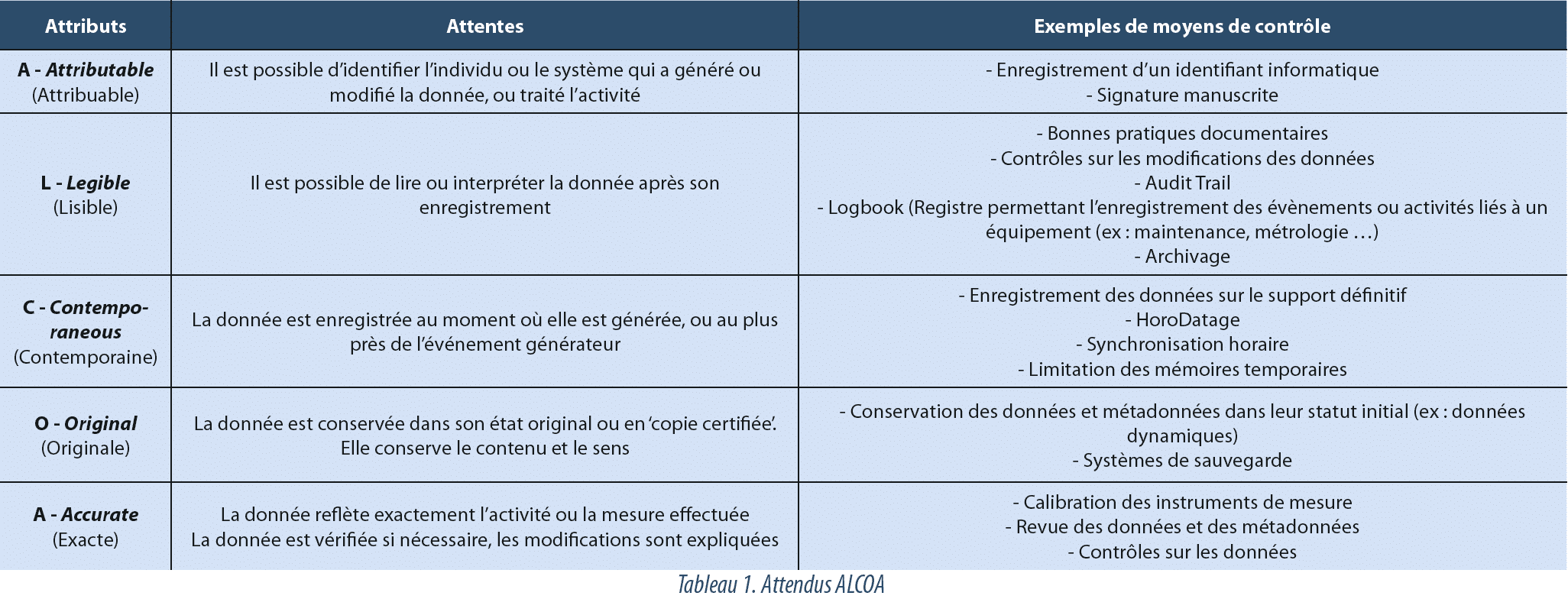
ALCOA
The authorities agree in considering thatData Integrity is covered when data comply with the ALCOA requirements (Attributable, Legible, Contemporaneous, Original and Accurate). This acronym summarizes the characteristics which demonstrate that the events to which data are subject have been correctly documented, and that they can be used to support a decision. The ALCOA+, concept, which provides additional specification by adding the terms Complete, Consistent, Enduring and Availableis also found in some publications.
Data Integrity is thus an essential requirement of the pharmaceutical quality system, and the main expectation is that the ALCOA principles are observed for all activities governed by GxP (see table 1).
Manufacturers, distributors and operators must therefore be able to detect faults, in the organization or systems, which may lead to the corruption of data, and result in an erroneous decision. This detection includes intentional and non-intentional corruption of data, and applies to electronic and paper media..
The different guidelines issued on Data Integrity have introduced a terminology which is important to assimilate, although some definitions can vary slightly from one text to another. Correct interpretation of some terms leads to better control of data integrity.
Metadata
Metadata are information connected to a data item, which add some context and allow its meaning to be better understood. The integrity of these metadata must be ensured. For example, the link between a data item and its time stamp (the date and time it was acquired) is always maintained, and this time stamp itself complies with ALCOAexpectations.
Static and dynamic records
Regulatory bodies, in particular the FDA, distinguish between static and dynamic records. Data in a static record are frozen, and have no reason to be modified. This is very often the case with a hard copy or an electronic image. A weigh ticket for example, does not generally need to be processed by a user to be exploitable and necessarily contains all the information needed to interpret the result.
This is not the case with a dynamic record format, which authorizes an interaction with a user for the purposes of its exploitation. On a chromatogram, for example, the integration parameters can be modified, a peak then appearing wider or narrower. The dynamic nature of a record must thus be preserved, in order to be able to retrieve the same result on the basis of the same data. In the case of the chromatogram, this signifies that printing after integration may not be sufficient to interpret a result, and that access to data stored electronically must be maintained.
Audit trail
The audit trail is a secure event log, tracing changes made to a system using time-stamping, generated by the system itself. The objective of the audit trail is to be able to reconstruct the events linked to any creation, modification or deletion of a critical data item.
The audit trail is considered metadata as it allows the “when”, “who”, “what” and “why” of a change to be known.
Backup and archiving
A backup is a copy of data, metadata and configuration parameters that is stored for the purposes of restoration in the event that the original data are lost.
Archiving is the long-term storage of data, for the purposes of being able to consult data throughout their retention period.
The integrity of backup and archived data, must be maintained.
Data Lifecycle
The Data Lifecycle is all the phases of the process by which data are recorded, processed, revised, reported, stored, recovered and subjected to review. It extends from the generation or acquisition of a data item or a data set, to their destruction or deletion.
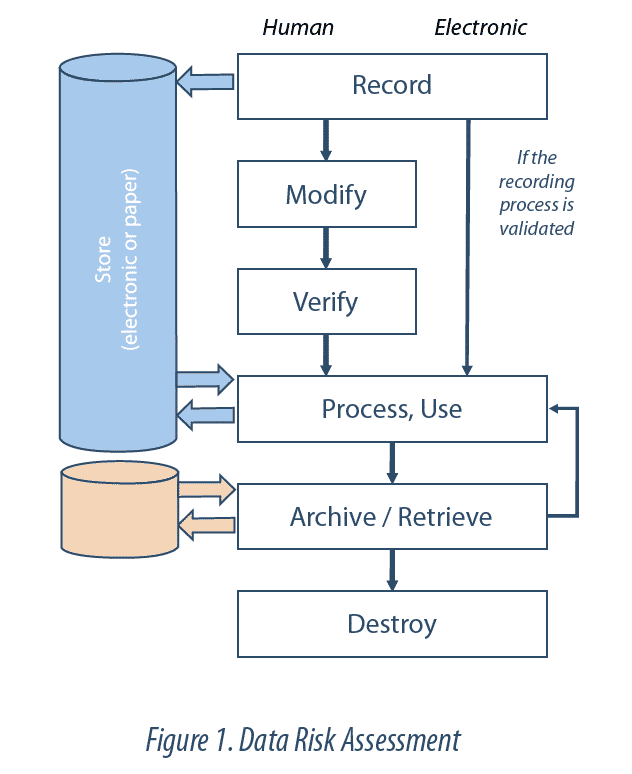
Data integrity must be guaranteed throughout their life cycle. This implies control of the Data Lifecycle. In other words, all steps associated with each data item or critical record are identified and understood (e.g. creation, storage, transfer, modification, archiving), so as to be able to detect any risk of modification or corruption. Review of the Data Lifecycle thus enables all manipulations applied to the data (e.g. calculations, exclusions) to be understood, and allows us to go back to the raw data. This review forms part of the Data Governancesystem.
Data Governance
Data Governance represents all the provisions which aim to ensure that data, in whatever format they are generated, are recorded, processed, stored and exploited in a way that guarantees a complete, consistent and accurate record throughout their life cycle.
Data Governance is an integral part of the pharmaceutical quality system and is based on the three lynchpins that are behavior, organization and technique.
Behavior that is aligned with the principles of Data Integrity implies in particular:
- an understanding of the importance of the subject by all staff involved (by means for example of a code of conduct, or a code of ethics),
- involvement of management,
- correct reporting and handling of deviations.
Organizational provisions may be, for example:
- a risk-based approach,
- the establishment of procedures,
- staff training,
- segregation of duties,
- review of data and routine checks,
- periodic reviews and surveillance of the system.
Technical systems involve, among other elements, the following subjects:
- the use of validated computerized or automated systems,
- the implementation of Audit Trails,
- safeguarding of the media on which the data are recorded,
- access control,
- the establishment of backup and archiving systems.
Review of the Data Governance system allows for an evaluation of whether there is a correct interaction between behavior, organizational measures and technical systems within departments.
As the issue of Data Integrity has for a long time been equated to a computer problem, technical systems are very often in place, whether these are security and control computer functionalities, or process automation, intended to limit errors associated with human interventions.
The introduction of technical solutions is not however enough if they are not operated in an appropriate and efficient manner. Theaudit trail, for example must be subject to review, to ensure that no uncontrolled modification has been carried out. This review is not applied exhaustively to all events that have arisen in the system but focuses on deviations observed in the operation of the data procurement process, and this only for critical data. The presence of an audit trail within a system does not therefore on its own guarantee the absence of controlled modifications, its operating rules must be defined and followed, such as the setting up of a systematic review procedure for certain events, or exceptionally the validated use of a review report.
The organizational aspect of Data Integrity supposes a certain maturity on the subject, with the full involvement of core activity staff and management, and an awareness of risks at company level, which sometimes requires a change of culture.
Data Governance follows a risk-based approach, to identify critical data (Data Criticality) and the associated risks of corruption (Data Risk), in order to adjust control efforts to just what is needed, in a balanced manner with other quality activities. The level of effort to be expended on data control is set in accordance with their criticality and their impact on CQA (Critical Quality Attributes) or their release data. Likewise, effort expended is graded in a manner proportional to the possibility of detecting data corruption. Finally, the efficacy of the provisions in place is managed and reviewed periodically.
Data Criticality
To determine the criticality of a data item, the following questions are asked:
- “Which decision do the data influence?”
- “What is the impact of the data on product quality or safety?”
The decisions influenced by data differ in importance, and the impact of data on a decision also varies. Effort is focused on the most critical data.
Data Risk
The degree of vulnerability of a data item to an uncontrolled modification (intentional or not), is assessed through a risk analysis. The factors to be taken into account in this analysis may be the complexity of the process analyzed, its level of automation or the subjectivity of the interpretation of results. The assessment is performed at the limits of the operating process, and not only functionalities or computer technologies, so as to take account of interactions with users or interfaces between systems, at each step of the life cycle.
Validation of computerized systems, while this is always necessary, is now no longer sufficient to ensure control of the risks associated with data integrity.
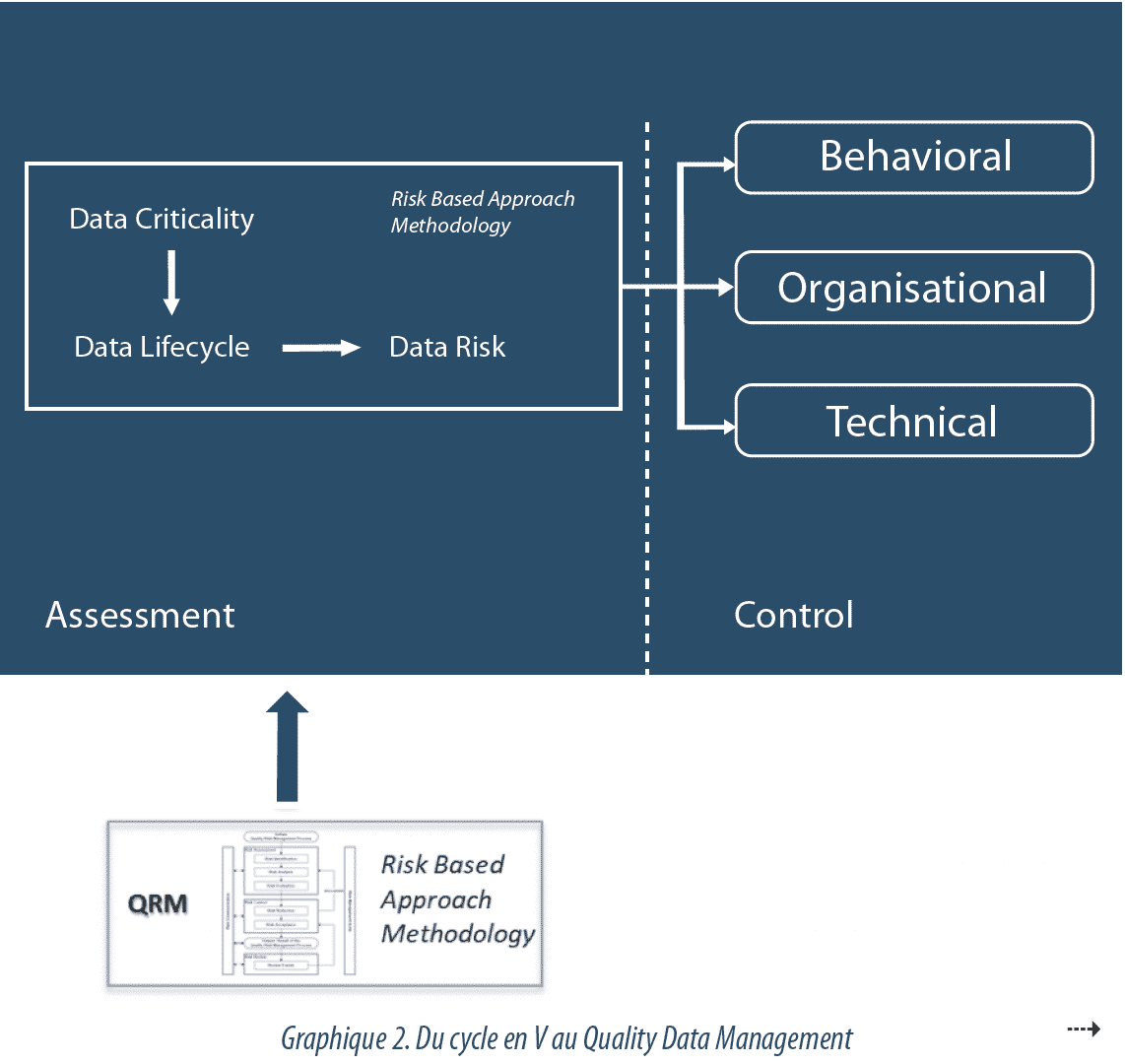
This is where the main change in the approach under consideration lies: the introduction of a Data Management system (see Graphic 2).
Data Management
Initially control of Data Integrity was based on the validation of computerized systems. Each system followed a V-model cycle, during which operations were defined, developed and tested, with system compliance being conferred by a validation report. Then the approach developed into one of risk control by integrating the system environment and its life cycle. This Risk-Based Approach, conveyed particularly by GAMP52 , included checks that various organizational aspects for exploitation were in place (training, procedures…), but remained at system level.
Today, the multiplication of computerized systems, very often interfacing with each other, and their high configuration level, correlated with the complexity of flows, make this approach obsolete. It is becoming necessary to move from a system-focused model to a data-focused model, with the setting up of a Quality Data Management system, structured and driven by risk, and ensuring consistency of processing within an organization. This model incorporates a continuous awareness of data integrity for all critical processes, regarding electronic data, paper data, interfaces between the two during the life cycle, and not only on critical systems.
Data Management is steered by a point of contact, the Quality Data Manager, reporting to the Quality Department, in contact with all departments that manage or manipulate critical data which support pharmaceutical decisions. This point of contact, who is the guarantor of the principles and rules of Data Integrity, directs their implementation, provides support for operating staff and keeps management informed of the greatest risks.
Conclusion
Good practices in Data Management and Data Integrity provide confidence in the robustness of pharmaceutical decisions and form an integral part of the quality system. Data corruption risks must be controlled in the same way as product risks, as the quality of a pharmaceutical product is closely linked to the quality of its traceability records.
Share article


