


Sommaire
- La restriction des PFAS dans l’industrie : enjeux réglementaires et impacts sur l’industrie pharmaceutique
- La technologie Blow-Fill-Seal dans l’industrie pharmaceutique : performance, applications et durabilité
- Key Allies in Preventing Contaminants and Impurities in Bioproduction
- Choosing the right vial: packaging sterile drug products with foresight
- Combination Products in the United States and European Union: Differences and proposed strategy to prepare common CTD Quality Module 3
- Blood plasma processing. When every drop counts
- L’analyse de la normalité en Continued Process Verification
- Qualification of impurities
- Pharma 2052
Qualification of impurities
Impurity has been defined by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) as any component of the new drug substance that is not the chemical entity defined as the new drug substance,
or any component of the new drug product that is
not the drug substance or an excipient in the drug product. Therefore, any molecule found in the drug substance or drug product and not identified as the drug substance itself or as an excipient is considered as an undesirable component matching with the definition of impurity. Such undesirable compounds resulting from manufacture or degradation of the drug substance or the drug product can fall into different categories of impurities, depending on their nature and origin.

Introduction
According to ICH regulatory guidelines, impurities in new drug substances are coming from the synthesis or storage of the drug substance and can be organic or inorganic. Likewise, impurities in new drug products result from the degradation of the drug product. Another category gathers all organic volatile chemicals that are used or produced in the manufacture of drug substances or excipients, or in the preparation of drug products. Such impurities are called resi- dual solvents. Finally, elemental impurities refer to all inorganic impurities coming from synthesis or degradation of new drug products. For each category w above, the ICH established internationally recognized regulatory guidelines to ensure quality of new pharmaceutical products and thus final patient’s safety. To this end, European, American, and Japanese authorities are particularly attentive to the regulatory compliance of dossiers when examinating applications for marketing authorisations as well as variations falling into the scope of such guidelines. In addition, new impurities appearing post-marketing as well as impurities overcoming acceptance criteria defined in specifications of already marketed products should be controlled. Pharmaceutical stakeholders are therefore concerned with adequate management of impurities including qualification of such extraneous compounds. Where in-house expertise or internal ressources are lacking, manufacturers are susceptible to externalise impurity toxicological qualification activities by referring to independent toxicology consulting specialists. Outsourced activities in qualification domain mainly include delegation of toxicological studies and strategy orientation. Depending both on the type of impurity and problem addressed, expert toxicologists can provide appropriate guidance in line with current regulatory requirements to manufacturers in need.
Part 1
As a case example, consider impurity A found in the drug product ABC containing the drug substance A’ and being commercialized for the treatment of retinal migraine in both adult and paediatric patients. Origin of impurity A has been elucidated by the internal pharmaceutical industry quality team and is arising from reaction of the drug substance with an excipient. According to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, such compound is classified among “Impurities in New Drug Products” for which ICH Q3B guideline applies. The ICH Topic Q3B (R2) Impurities in New Drug Products specifies the need for reporting, identifying, and qualifying impurities based on pre-defined thresholds and provides guidance on the way to conduct these assessments (Figure 1). Back to our example, consider that impurity A represents 0.85% of the drug substance and suppose that the maximum daily dose (MDD) of A’ drug substance is 2 g. In line with reporting and identification thresholds defined by ICH Q3B, impurity A has been adequately reported in the registration application and identified as substance S. Thereafter, A’ falling into the category of a MDD less than or equal to 1 g/day and regarding the 0.85% level of impurity A, qualification process for this impurity should have been considered.
External toxicologists are usually invited to intervene at this stage of the process.
When a pharmaceutical industry refers to independent specialists for guidance on this aspect, generally the initial step is to request for any experimental data the Sponsor may have on the impurity concerned. In most cases, there is no experimental results available. If so, an extended bibliographic search, screening most of reliable international databases, is proposed. As an example for impurity A, we assume that sufficient data has been collected to appropriately qualify the compound. Afterwards and in regard to the data selected, a monography is performed, from which a safety limit can be established based on a conservative approach. Generally, toxicologists are able to calculate a permitted daily exposure (PDE) corresponding to the daily quantity of substance below which no adverse effects are expected in susceptible individuals following exposure for a lifetime by the indicated route. Based on a worst-case scenario, the most susceptible population must be taken into account.
For example, consider product ABC is indicated from 5 years of age, therefore the PDE should be determined based on the weight of a 5-year individual. Once the PDE obtained, the objective is to perform a complete toxicological risk assessment to finally qualify the impurity. Consider example of impurity A. After having collected all published data on the substance, a PDE of 25 mg/day could be established for adults and children over 5 years old. This value needs now to be compared to the maximum exposure of patients to impurity A in order to characterise the risk. For estimation of exposure to impurity A, the first step is to determine the maximum daily dose of the drug product and thereby deduce the maximum daily dose of impurity A. Based on the maximum exposure to impurity A on the one hand, and the PDE value on the other hand, a risk characterisation ratio has been calculated as follows:
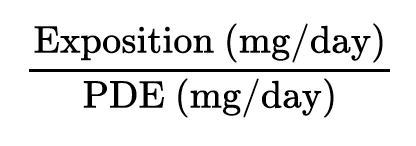

For impurity A, given the PDE of 25 mg/ day and an estimated maximum expo- sure of 17 mg/day, the following risk ratio could be determined:
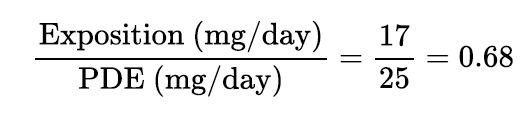
The resulting ratio being below 1, the health risk associated with impurity A is controlled. Thus, qualification step is now completed and impurity A is considered adequately qualified.
In addition, most of the time toxicologists are requested to find out the maximum acceptable amount of impurity in the drug substance or drug product. In our example, impurity A administered up to 25 mg per day is regarded as a safe situa- tion. In percentage terms for impurity A, this represents a maximum acceptable concentration of 1.25% in the drug subs- tance. In conclusion, the pharmaceutical industry is advised to control impurity A in A’ drug substance below the safety limit of 1.25% of A’ drug substance.
Part 2
Another example to illustrate the safety assessment of impurities within the framework of international recommendations would be impurity B. Impurity B is a by-product produced by chemical synthesis of B’ drug substance. The corresponding drug product, BCD, is under development and indicated for the treatment of severe acne in adult patients with a maximum treatment duration of 12 months. According to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, such substance falls into “Impurities in New Drug Substances” category for which ICH Q3A guideline applies. As impurity B belongs to organic impurities, reporting, identification, and qualification activities should be considered in the same way as for impurity A, the only difference being slightly lower thresholds. Another strategy would have been followed if impurity B had been inorganic, indeed for such compounds established thresholds do not apply. Back to our example, suppose that the maximum daily dose of B’ drug substance is 3g. Impurity B being recognized as a potential impurity most likely to arise during drug substance manufacturing with an anticipated level of 0.2%, reporting is deemed necessary by ICH Q3A. Once adequately reported and in conformity with the ICH, impurity B is identified by the manufacturer although no CAS number can be attributed. Afterwards, qualification process is required since impurity B is present at a level of 0.2%, thus surpassing the corresponding qualification threshold (Figure 2).
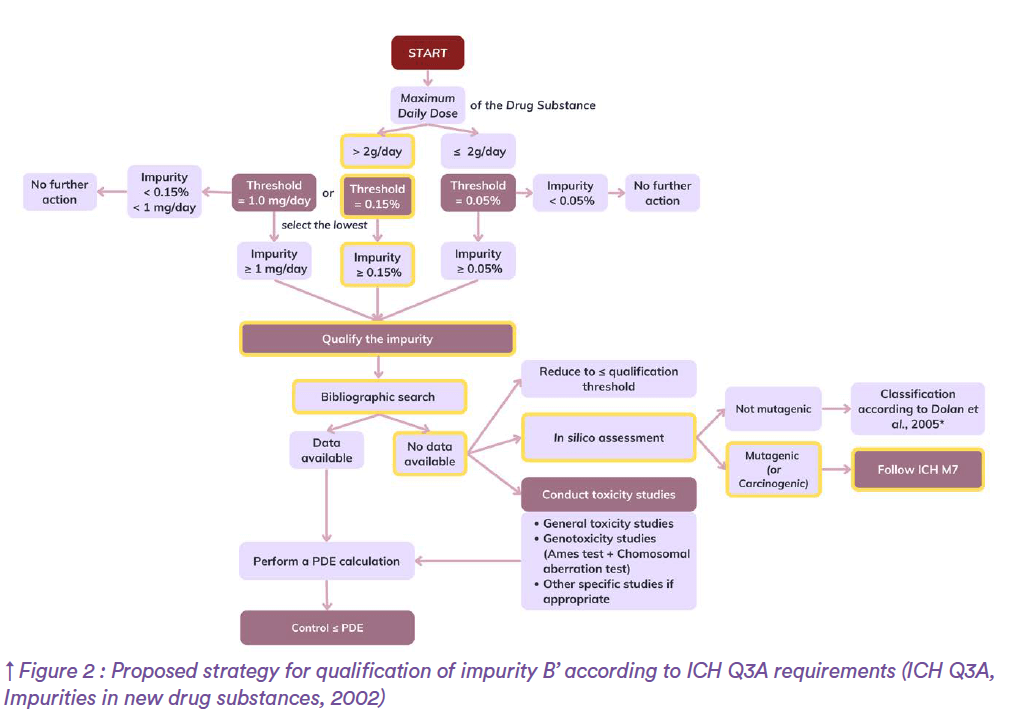
At this stage of the process, when there is no experimental data available internally, independent toxicologists are used to suggest an extensive bibliographic search. In the present example, consider that no data could be found. In such cases, a number of options can be proposed by qualification specialists.
The first one would be to merely control impurity B below the qualification threshold of 0.15%. For the manufacturer, such choice might involve adjustment of the drug substance manufacture process, that could be very expensive or simply not feasible due to technical issues.
In the absence of bibliographic references, and if controlling below the qualification limit is not considered by the industry, performing a battery of toxicological studies may constitute another way to collect data so as to establish a PDE. Using this PDE value, toxicologists become then able to conduct a risk assessment and qualify the impurity on the same basis as for impurity A. Significant cost of such general toxicity and genotoxicity studies in terms of time and budget constitutes the major disadvantage of the option.
Finally, last alternative would be performing an in silico toxicological method using a conservative approach. If in silico results demonstrate a potential for mutagenicity, then ICH M7 should be followed. If there is no potential for mutagenicity, then thresholds from Dolan et al.1 may apply depending on structural alerts identified.
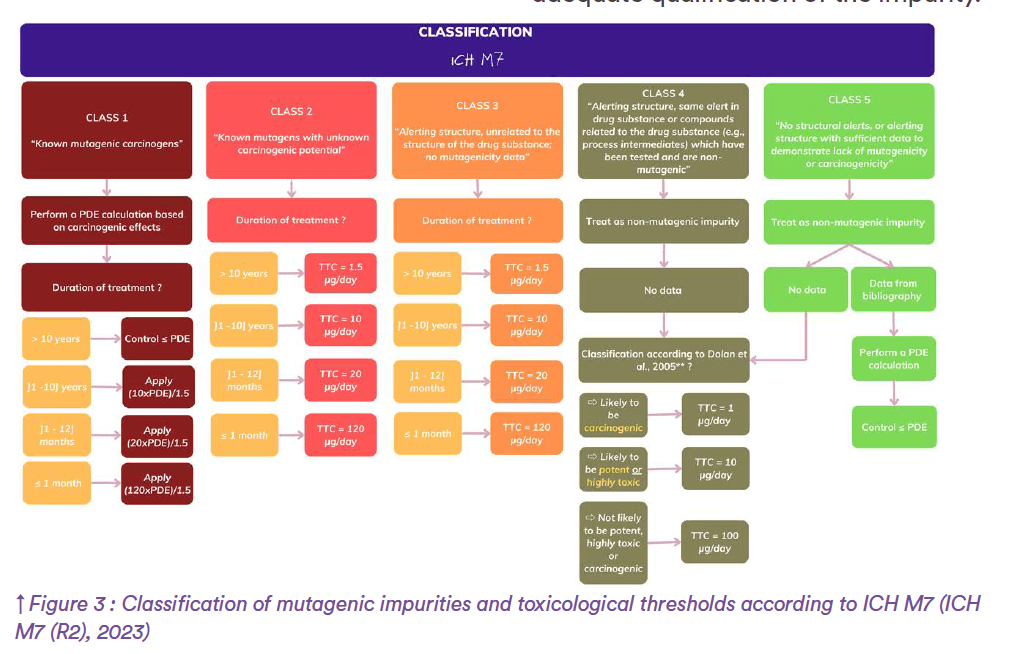
Now, if we go back to the above example and consider that the industry selects the last option, external in silico experts are invited to launch two (Q)SAR models based on the structure of impurity B identified earlier. To this end, complementary expert rule-based and statistical-based methods are recommended to generate relevant alerts. Following in silico assessment, suppose that predictions demonstrate a potential for mutagenicity. Henceforth, toxicologists are encouraged to refer to the ICH M7 Guideline on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk for qualification of impurity B. As per ICH M7, substances can be classified into one of the following classes (Figure 3):
– Class 1 “Known mutagenic carcinogens”
– Class 2 “Known mutagens with unknown carcinogenic potential”
– Class 3 “Alerting structure, unrelated to the structure of the drug subs- tance; no mutagenicity data”
– Class 4 “Alerting structure, same alert in drug substance or compounds related to the drug substance (e.g., process intermediates) which have been tested and are non- mutagenic”
– Class 5 “No structural alerts, or alerting structure with sufficient data to demonstrate lack of mutagenicity or carcinogenicity”
When in silico assessment reveals alerting structures for mutagenicity as happened for impurity B, ICH M7 recommends checking whether the same alert appears with the drug substance. For impurity B, suppose that the same alerting structure is observed while B’ drug substance is screened. As part of the preclinical drug development strategy, the industry concerned reports that B’ has already been tested in Ames test and in vitro micronucleus assay, showing positive results for mutagenicity. Therefore, impurity B is considered as a known mutagen with unknown carcinogenic potential, thus falling into class-2 compounds. In such cases, a Threshold of Toxicological Concern (TTC) is defined according to ICH M7 requirements.
The TTC corresponds to a level of daily exposure to a chemical over a lifetime that is considered to be of no appreciable risk to human health. Thus, such method provides an estimate of safe exposure for any mutagenic compound and is recognized as very conservative with a theoretical excess cancer risk of <1 in 100,000.
A TTC of 1.5 μg per day has been established for mutagenic impurities occur- ring in new drug substances. However, adaptation of this TTC value is allowed by regulatory authorities when exposure to the impurity is not expected to last a lifetime. Back to impurity B, considering the maximum treatment duration between 1 and 12 months, a TTC of 20 μg/ day instead of the traditional 1.5 μg/day can be established according to ICH M7. Finally, expert toxicologists recommends controlling substance B below 20 μg/ day in BCD drug product so as to ensure adequate qualification of the impurity.
Conclusion
To properly ensure patients’ safety, phar- maceutical laboratories are strongly encouraged to follow ICH recommen- dations when evaluating real or potential impurities occurring in new drug subs- tances or drug products. The toxicolo- gical assessment of such undesirable compounds is a stepwise procedure with thresholds defining the need for repor- ting, identification, and qualification of impurities. When compound quali- fication is deemed necessary, several approaches can be proposed by inde- pendent experts as well as related toxi- cological services to support industries in the definition of safe limits. External toxi- cologists thus provide guidance on the best strategy to adopt, which sometimes requires “thinking out of the box” mode to establish appropriate safety thresholds with regards to client needs, industrial feasibility, and economic constraints. At the same time, independence of such experts allowing priorisation of patients’ safety may be regarded as side benefits of externalisation processes.
Reference
1. Dolan DG, Naumann BD, Sargent EV, Maier
A, Dourson M. Application of the threshold of toxicological concern concept to pharmaceutical manufacturing operations. Regul Toxicol Pharmacol. 2005, 43(1), 1-9
Partager l’article

