


Sommaire
- Considerations for Validating Aseptic Manufacturing Processes.
- Simulation des procédés de fabrication aseptique de suspensions de nanoparticules ou microsphères : Challenges & facteurs de succès au travers d’un retour d’expérience.
- L’isolateur jetable pour le remplissage aseptique, innovation ou hérésie ?
- A3P/AFI Survey on Sampling & Testing Practices for In-Process Pre-Filtration Bioburden for Sterile Products: Presentation of the Results & Critical Discussion.
- Projet de norme internationale amené à être amendé PR ISO 11737-3 : Stérilisation des produits de santé - Méthodes microbiologiques - Partie 3 : essai des endotoxines bactériennes.
- Real-time detection of CFU growth with the ScanStation® smart incubator expedites the environmental monitoring process.
- MTP: What It Is, Why It Matters. Module type package standard is a major step forward for plug-and-produce manufacturing.
- How Pemflow / D.O.C. manage Extractables and Leachables Qualification of the in-process materials and primary packaging.
Considerations for Validating Aseptic Manufacturing Processes.
In contrast to manufacturing process validation (PV), aseptic processing validation demonstrates a low level of contamination risk associated with the aseptic process by simulating the manufacturing process using microbiological growth media in place of the intended drug solution. The use of media for this purpose is known as Aseptic Process Simulation (APS) or “media fills”[1], and regulators require demonstration of the aseptic capability of such processes, where terminal sterilization cannot be used.

A successful APS program has four requirements:
- Significant operator training, skills, and supervision;
- Effective maintenance, cleaning, and disinfection;
- Operational oversight by quality assurance; and
- Microbiological monitoring. Sterility assurance in aseptic processing requires all contributing elements to be qualified or validated—such as the heating ventilation and air conditioning (HVAC) systems, cleanroom environment, material transfer, equipment, and manufacturing process steps, including sterilization processes and sterilizing filtration—and for operating personnel to be trained and qualified.
An APS program consists of a minimum of three successful initial media simulations, followed by repeat media simulations at six-monthly intervals. Any media fill failures require thorough investigation and root cause analysis; further media simulations may be required.
Aseptic manufacturing is typically carried out in conventional cleanrooms with container filling and stoppering in Grade A laminar airflow (LAF) in a Grade B background environment. The filling environment may be further protected by a restricted-access barrier system (RABS) with glove ports to access the filling line, or processing equipment for critical steps may be enclosed in glove boxes or isolators. Each of these enhances the sterility assurance of the filling process and presents challenges for material transfer, operator access, environmental monitoring, and APS.
1. Overview of Global Regulations
Aseptic manufacturing and validation follow current GMPs and related GMP Annexes or guidance documents. The regulations govern the validation, manufacture, and control of sterile products for injection (also eye drops and advanced therapy medicinal products). Current guidelines[2-10] come from the European Union/Pharmaceutical Inspection Convention (EU/PICS), the United States Food and Drug Administration (US FDA), and the World Health Organisation (WHO) and may be inspired by the International Organisation for Standardization (ISO) and the Parenteral Drug Association (PDA) monographs[11–14], such as those relating to cleanroom air-cleanliness classification[13] and particle monitoring[14].
Parenteral drug products are assessed as having a high safety risk profile; thus, the protocols, results, and reports for APS form an integral part of regulatory submissions such as investigational new drug (IND) applications, new drug applications (NDAs), and marketing authorizations (MAs). GMP documents such as training records, environmental monitoring reports, deviations and investigations, and qualification of facilities, equipment, and utilities may be scrutinized during regulatory inspections.
APS must achieve three consecutive media fill batches meeting target acceptance criteria. The solution filtration process must be validated against a microbial challenge with 107 colony-forming units per square centimeter of the filter medium of Brevundimonas diminuta, a small-celled Gram-negative bacterium to be suspended in the drug solution. Media fill batch sizes and acceptance criteria incorporated in GMP Annex[3] and Guidance[7, 8] include:
- Filling less than 5,000 units, zero contaminated units should be detected. A contaminated unit is considered a cause for revalidation following an investigation.
- Filling 5,000 to 10,000 units; one contaminated unit should lead to an investigation, including consideration of a repeat media fill. Following the investigation, two or more contaminated units are cause for revalidation.
- Filling more than 10,000 units; one contaminated unit should lead to an investigation, and two or more contaminated units are cause for revalidation.
Regulatory inspections in aseptic manufacturing companies have increased, and the number of monoclonal antibody and advanced therapy medicinal products requiring aseptic filling has grown.
Some typical examples of GMP failures and APS issues that have appeared in warning letters and summaries by regulators are as follows:
“Inadequate Smoke Studies
Our inspection found that you lacked smoke studies to evaluate whether unidirectional airflow exists on your (b)(4) ointment aseptic processing line.
Inadequate Media Fills
Your media fill program was inadequate. Simulations were not performed at a sufficient frequency and were not representative of worst-case production conditions.” FDA Warning Letter 320-17-48 dated August 31, 2017
Other typical audit observations related to media fills are shown below :
- Media Fill Design
- Fill and incubate insufficient vials
- Incorrect incubation conditions or duration
- Failure to carry out adequate growth promotion testing of media batches
- Lack of evaluation by smoke studies on the effects of interventions on unidirectional (laminar) airflow
- Interventions in media fill not representative of actual process interventions by operators
- Operational
- Failure to reconcile and incubate all integral media filled vials
- Media fill is not performed after significant activities such as major facility shutdowns that may compromise cleanroom state of control.
- Media Fill Failure Investigations
- Failure to identify the microorganisms in contaminated media units to species level.
- Thorough investigation not performed to determine the root cause prior to repeating media fill.
- Operator behavior
- Poor aseptic technique such as rapid movement in critical areas, and failure to sanitize gloved hands periodically.
- Inadequate training of media vial inspectors to examine media-filled units following incubation.
2. Points to consider for media fills
The following points should be considered when designing the media fill study for an aseptic manufacturing process.
Worst-Case Approach in APS
The aseptic manufacturing process should involve a “worst-case” approach as a challenge to the robustness of the aseptic operations. Risk assessment principles should be used to determine the worst- case challenges related to line speed, container size, batch size, holding times, configurations, and operating conditions.
Examples of worst-case challenges:
Operating conditions
- Maximum number of personnel in aseptic area
- Shift changes, personnel changes, and operator breaks – Equipment/room clean hold time
- Equipment sterilization hold time
Filling process
- Aseptic assembly of equipment and aseptic connections prior to filling
- Slowest filling speed with widest opening vials/containers
- Maximum fill volume for small vials/containers
- Maximum batch filling duration (including lyophilizer loading and door opening duration)
- Operator fatigue as contamination risk
Process Interventions
The regulatory expectation is that interventions included in APS must be compliant with current GMPs, and APS must not be used to justify poor aseptic practice or equipment design.
Routine interventions should be performed as per standard operating procedures or batch records. They may include charging stopper and seal hoppers, removing jammed stoppers or toppled vials, and collecting samples for environmental monitoring or in-process control. Interventions to be followed in the event of machine jams and spills may include partial line clearances, including removal of exposed units.
Nonroutine interventions may include changing filling needles or unexpected events such as breakdown maintenance, line stoppages, machine adjustments, and material transfers. Interventions may be grouped by access point, and their risk assessed so that worst-case (highest risk) interventions are included in the study.
Simulation of Lyophilization in APS
EudraLex Annex 1 (2009)[3] states, “The process simulation test should imitate as closely as possible the routine aseptic manufacturing process…”. It is unlikely that a product lyophilization cycle can be replicated during media simulations due to the constraint of maintaining the media’s ability to support microbial growth; deviation from the production cycle must be justified in the protocol.
Based on risk analysis, the aeration or vacuum-break step in the lyophilization cycle may have a higher risk of contamination because of turbulence[15] and the possibility of entrained particles entering the containers. As the application of full vacuum is not possible during APS, multiple partial vacuum steps should be considered to simulate the worst-case aeration. The media volume in the vials before lyophilization must ensure that the wetted surface of the container mimics the production case.
Media simulation of lyophilization should involve loading the required number of media-filled vials as per routine production procedures. An important consideration is to ensure that the duration the lyophilizer door is open to the cleanroom environment is at least the maximum time incurred when loading a production batch.
At the end of the lyophilization cycle in APS, sterile-filtered compressed air should be used to break the chamber vacuum to avoid inhibiting microbial recovery and growth in the stoppered vials. Nitrogen gas is used to break the vacuum only if an anaerobic media simulation is undertaken.
Incubation and Inspection
Visual inspection is performed for APS batches after filling, stoppering and sealing to identify defects such as visible foreign matter, high or low fill volumes, and damaged vials, stoppers, or seals. These defective units would normally be removed (rejected) from product batches, but all defective units must be retained and all integral containers incubated in APS batches. Non-integral (broken or otherwise damaged) containers must be recorded and reconciled with the batch quantities. The incubation conditions are selected to be optimal for recovery and allow for detection of both slow-growing and normal contaminating organisms, i.e., to detect microorganisms that might otherwise be difficult to culture. The incubation conditions used generally are 20°C to 25°C for seven days (lower temperature first) followed by 30°C to 35°C for an additional seven days.
Containers during incubation must be turned to ensure that growth medium contacts the entire interior surfaces. Chart printouts or electronic records of the incubation conditions must be maintained, including the date and time of incubation commencement, turning of vials, transfer to the second incubator, and further turning and completion of incubation. Incubated vials must be inspected by operators qualified to distinguish sterile vials (“no growth”) from vials showing microbial growth (surface pellicle or turbidity in the solution). A small number of filled vials with no microbial growth should be selected for use as “after-test” growth controls.
Microbiological Growth Medium
Media for microbial recovery and growth are defined in pharmacopoeias—such as the United States (USP), European (Ph. Eur.), Chinese (ChP), and Japanese (JP) Pharmacopoeia—and should be made, sterilized, and used for media fills as per the manufacturer’s instructions. Generally, tryptone soya broth (TSB) or equivalent, which supports the recovery and growth of viable aerobic microorganisms, is used. Special circumstances may require considering anaerobic growth media such as fluid thioglycolate medium (FTM), which supports the recovery and growth of obligate or facultative anaerobic bacteria. The growth medium, supplied as a powder, is a critical material for APS.
The manufacturer is recommended to be qualified and monitored as an approved supplier; growth promotion certificates may be obtained with each media powder batch. Before release, batches of the media for APS should be reconstituted, sterilized, and subjected to quality control for growth promotion by inoculating with ≤100 colony-forming units (CFUs) of representative compendial strains of microorganisms (a strain from environmental monitoring may be included).
Environmental Monitoring during Media Fills
All routine and normal processes (such as cleaning, disinfection, and maintenance) should maintain the cleanroom environment in its qualified status. Maintenance includes particulate and microbiological environmental monitoring to demonstrate that the specified cleanroom environment conditions are maintained. Monitoring results may also provide key information for investigating a failed media fill.
Particulate monitoring consists of continuous monitoring for particulates in the <0.5 μm and <5.0 μm ranges, using a particle sampler attached to an isokinetic probe located near the fill point in the Grade A area. A permanent record of the particle counter’s printout (or certified true copy if the printout is on thermal paper) must be attached to the APS or product batch record. The regulatory/ action limits per m3 air volume are not more than 3,520 particles in the <0.5 μm particle size range and not more than 20 particles in the <5.0 μm range.
Each batch of environmental sampling plates must be tested for sterility and growth promotion capability against the recommended compendial strains of microorganisms before releasing for use. The microbiological methods should include a map of the locations where samples are to be taken or plates exposed. The methods are as stated in EudraLex, current Annex 1[3]; active air sampling (1 m3 sample volume) onto 90 mm agar plates; settling plates 90 mm in diameter with exposure up to four hours (if the APS or production filling lasts longer, new settling plates must be exposed for subsequent four- hour periods); surface contact plates, 55 mm in diameter, which are contacted against cleanroom walls, floors, or operator gowns; and finger plate samples, performed by cleanroom operators during the filling period and upon leaving the cleanroom (four fingers and thumb onto the surface of a 90 mm TSA settle plate).
Media are usually tryptone soya agar (TSA) for viable aerobes and sabouraud dextrose agar (SDA) for fungi (molds) and yeasts. Surface contact plates may be TSA, incorporating a neutralizing agent to counter detergent residues from surfaces – agar residues are removed from the sampled locations by wiping with 70% alcohol.
The regulatory action limits for the microbiological monitoring results of the Grade A cleanroom areas (Grade A LAF in Grade B background, RABS, or isolator) in CFUs, are as tabulated in EudraLex, current Annex 1[3]. Adjacent Grade B, C, or D cleanrooms through which operator gowning and material transfer for APS occur should also be monitored; the stated regulatory limits for these cleanroom grades are also included in EudraLex, current Annex 1[3]. The frequency of monitoring Grade C and D cleanrooms is determined based on quality risk assessment; however, such monitoring at the time of a media fill may assist the investigation of a discrepancy or failure.
For media fills, the number of CFUs on the environmental monitoring plates in Grade A (LAF, RABS, or isolator) and Grade B areas must be enumerated and identified to species using available biochemical or nucleic acid identification methods. The isolates can be compared with organisms in any contaminated units. Information regarding the numbers, species, and locations of contaminating microorganisms may prove crucial in investigating a failed media fill (also from monitoring of Grade C and D areas).
Operator Training and Qualification
Staff qualified to work in aseptic manufacturing, including maintenance and quality personnel, must participate in media fills.
Operators performing APS and aseptic manufacture must be trained in relevant procedures, including cleanroom gowning and aseptic behaviour (as well as product-specific procedures):
- After initial theoretical training, aseptic operators should be allowed to practice their manipulations in a mock-up or nonsterile practice environment before participating in cleanroom operations.
- Sterile materials and equipment are handled only with sterilized instruments, such as forceps. Between uses, instruments should be protected from contamination.
- After initial gowning, sterile gloves are regularly sanitized by spraying with a qualified sanitizing agent such as sterile 70% isopropyl alcohol (IPA) to minimize the risk of contamination. Personnel should not directly contact sterile products, containers, components, or critical surfaces.
- Rapid movements create turbulence, threatening the integrity of sterile environments and entraining particles. Operator movements should be slow and deliberate and should not disrupt LAF designed to protect critical processes.
- When performing aseptic manipulations (such as making aseptic connections, removing samples, or retrieving fallen or jammed components from a filling line), operators should be trained to approach the location slowly and deliberately from the side to avoid disrupting LAF.
3. Media Fill Failures – Root Cause Determination and Remediation
A critical step in the investigation is identifying species of the microorganism(s) in positive media vials, and of any colonies appearing on environmental monitoring plates, particularly those from the Grade A/B environments, including from RABS/Isolator monitoring. Identifying from colonies on plates exposed in the lower-Grade adjacent cleanrooms is also crucial, as materials or personnel have accessed the filling rooms through these lower grade cleanrooms. The ability to match the identification of an environmental isolate from the current (or recent) batch with the identity of the contaminating organism in the failed media units provides important clues to the root cause determination.
The review of the deviation should encompass the preparation and manufacturing processes, including cleaning and disinfection of the cleanrooms, sanitization/sterilization, and transfer processes for components and materials, HVAC and cleanroom operating parameters during the filling period, operation of the filtration process, and integrity tests and filling, stoppering and capping equipment, and taking and transfer of in-process or environmental samples. The review should focus on documentation, including any deviations or atypical events, and documented interviews with operators. The review should also include recent engineering work prior to media-fill batches. The key is to detect any significant processing discrepancy or error and equipment failure.
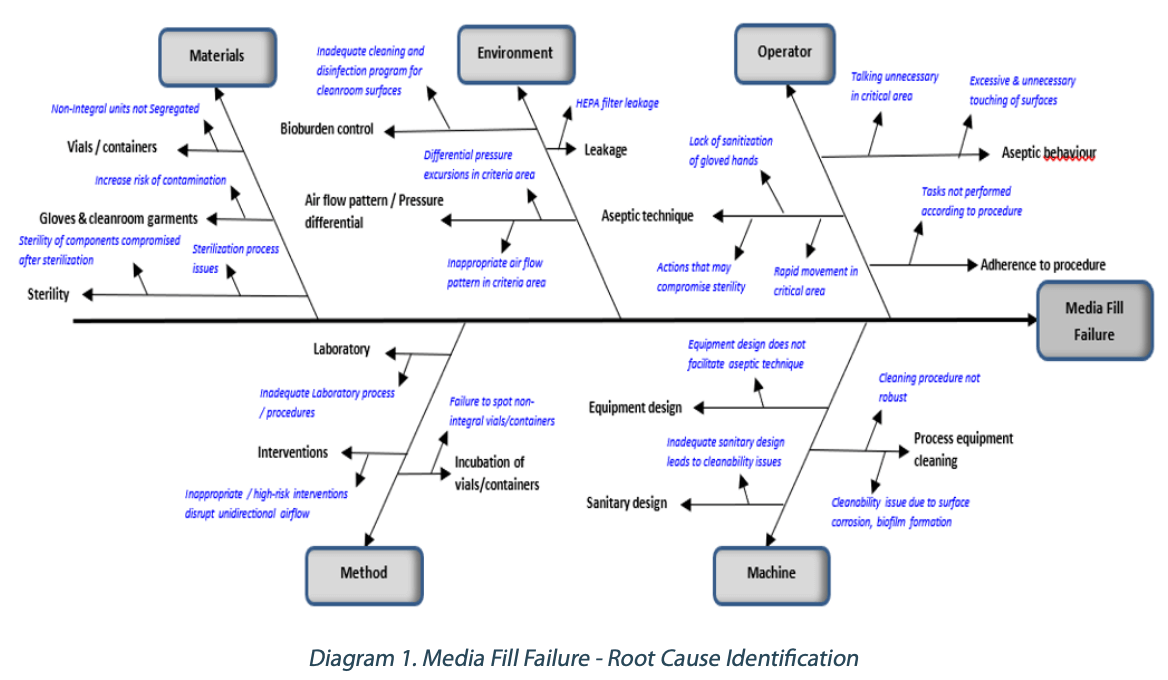
A fishbone diagram can be a valuable tool to investigate media fill failures and help identify the potential causes. Diagram 1 below shows an example of the fishbone diagram utilized.
Based on the potential failure modes identified in Diagram 1, the corresponding risk mitigation measures for the respective possible failure mode can be determined.
Operators play an essential role in ensuring the sterility of products is not compromised. Aseptic behavior, aseptic technique, and compliance to standard operating procedures are critical in aseptic manufacturing processes. Risk mitigation typically involves periodic aseptic behavior and aseptic technique training. Audits can also be conducted periodically to ensure strict adherence to the procedures. Equipment design that does not facilitate aseptic interventions can increase the risk of contamination during an intervention. Inadequate sanitary design in process equipment can lead to cleanability issues. Design qualification (DQ) and modifications may be required to mitigate the risk.
Process equipment cleaning needs to have a robust cleaning procedure considering the type of soil, cleaning parameters, and the use of an appropriate cleaning detergent if required. Periodic maintenance of stainless-steel surfaces can minimize the risk of corrosion, which can cause cleanability issues that can also lead to biofilm formation. Designing a robust cleaning and disinfection program utilizing sanitizers, disinfectants, and sporicidal agents is critical for bioburden control. Inappropriate air flow patterns in critical areas need to be remediated to minimize risk of contamination to products. Periodic maintenance and leak tests of the HEPA filters in critical areas ensure the integrity of the air filtration system.
Review and remediate laboratory procedures to minimize errors. High- risk interventions disrupt unidirectional (laminar) airflow and increase the risk of contamination. Smoke studies should be conducted and evaluated for risk of contamination (turbulence) for each intervention. Train inspectors to spot and segregate non-integral vials/containers so as to avoid incubation and causing media fill failure.
Verify sterilization processes meet acceptance criteria, and any deviation requires impact assessment. Contamination of sterile parts/ packaging components can occur after the sterilization process and during storage prior to use if there are minimal protective measures in place. Ensure these components are protected from contamination after sterilization by using an appropriate protective barrier.
Conclusion
The objective of the aseptic process simulation is to assess the likelihood of microbial contamination of the product during the aseptic manufacturing process. A critical aspect of designing a robust APS involves understanding the regulatory expectations and using risk assessment tools in the worst-case challenge design, including representative process interventions. Equally important are sound and effective programs for cleaning and disinfection of clean rooms, for equipment cleaning and maintenance, and for validation of sterilization processes. Competent operators and support staff, well-trained in aseptic techniques and behavior, with adequate knowledge of microbiology and deviation- and failure-investigation skills, should ensure compliance with CGMP regulations, and a successful APS program.
Partager l’article

Références
1. Chai, R and Barber D, Pharmaceutical “Validation of Aseptic Processes”, Pharmaceutical Engineering, Volume 42 No. 2, March-April 2022.
2. European Commission. “EudraLex, Volume 4: EU Guidelines for GMPs for Medicinal Products for Human and Veterinary Use. Part 4: EU Guidelines for Good Manufacturing Practice (GMP) Specific to Advanced Therapy Medicinal Products.” November 2017. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2017_11_22_guidelines_gmp_for_atmps.pdf
3. European Commission. “EudraLex, Volume 4: EU Guidelines for GMPs for Medicinal Products for Human and Veterinary Use. Annex 1: Manufacture of Sterile Medicinal Products.” Published 2008. https://ec.europa.eu/health/sites/default/files/files/gmp/2017_12_pc_annex1_consultation_document.pdf
4. European Commission. “EudraLex, Volume 4: EU Guidelines for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use. Annex 13: Investigational Medicinal Products.” Published 2017. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2009_06_annex13.pdf
5. US 21 Code of Federal Regulations Part 210 Current GMP in Manufacturing, Processing, Packing or Holding of Drugs; General. Updated September 13, 2021. https://www.ecfr. gov/current/title-21/chapter-I/subchapter-C/part-210
6. US 21 Code of Federal Regulations Part 211 Current GMP for Finished Drug Products. Updated September 14, 2021. https://www.ecfr.gov/current/title 21/chapter-I/ subchapter-C/part-211
7. US Food and Drug Administration. “Guidance for Industry. Sterile Drug Products Produced by Aseptic Processing— Current Good Manufacturing Practice.”September 2004. https://www.fda.gov/media/71026/download
8. WHO Expert Committee on Specifications for Pharmaceutical Preparations. Annex 2 WHO Good manufacturing practices for pharmaceutical industries; Main Principles. Published 2014.
9. WHO Expert Committee on Biological Standardization. Annex 4 General requirements for sterility of biological substances. Published 1973.
10. Pharmaceutical Inspection Convention/Pharmaceutical Inspection Co-Operation Scheme. “Recommendation on the Validation of Aseptic Processes.” Published January 2011. https://picscheme.org/docview/3446
11. PDA Technical Report #22,” Process Simulation for Aseptically Filled Products.” Revised 2011.
12. ISO Standard 13408-1. “Aseptic Processing of Health Care Products – Part 1: General Requirements.” Published June 2008. https://www.iso.org/standard/37842.html
13. ISO Standard 14664-1:2015. “Cleanrooms and Associated Controlled Environments — Part 1 Classification of Air Cleanliness by Particle Concentration.” Published 2015. https://www.iso.org/obp/ui/#iso:std:iso:14644:-1:ed- 2:v1:en
14. ISO Standard 14664-1:2015. “Cleanrooms and Associated Controlled Environments — Part 2: Monitoring to Provide Evidence of Cleanroom Performance Related to Air Cleanliness by Particle Concentration.” Published 2015. https://www.iso.org/obp/ui/#iso:std:iso:14644:-2:ed- 2:v1:en
15. Hamilton, D., T. Tharp, O. McGarvey, M. Frei, M. Dekner, S.B. Mehta, X. Chen, N. Zinfollino, S. Schneid, J. Briggs, M. Schreyer, and D. Hill. “A Better Approach to Aseptic Process Simulation For Lyophilized Products” Outsourced Pharma Published May 9, 2021. https://www.outsourcedpharma. com/doc/a-better-approach-to-aseptic-process-simulation- for-lyophilized-products-0001

Richard CHAI – STERIS
Voir le profil sur ![]()

David BARBER – STERIS
Voir le profil sur ![]()
