


Summary
- ASTM E2500 : let's cross the Bridge !
- From study of the context to use of appropriate water treatment technologies
- A new water management strategy for the pharmaceutical industry
- Continuous measurement in water for pharmaceutical use (WPU)
- Candida auris, an emerging pathogenic “killer” species
- Cleaning and disinfection program part of the lifecycle approach: a risk based rather arbitrarily imposed approach
- Pharmaceutical water: the inside story of installation maintenance
- Pharma & the New Healthcare Economy. Ready or not ?
ASTM E2500 is a standard relating to the specification, design and verification of Pharmaceutical and Biotechnological manufacturing systems and equipment. It is applicable to equipment; systems/processes; utilities (industrial, clean); laboratory systems; I.T. and automation systems that may affect product quality and patient safety.
“ASTM E2500 is first and foremost a mindset, a culture change!”
A consensus standard approved in May 2007, published in June 2007, and re-approved in 2012, this guide is in line with international regulatory requirementsparticularly ICH Q8, ICH Q9, ICH Q10 (Pharmaceutical Development/”Quality by Design”, Quality Risk Management, Pharmaceutical Quality System). Supported by the FDA, this approach, widely used in the USA, is beginning to be rolled out in Europe, particularly by manufacturersseeking to rationalize their system test phases prior to operational scale-up and thus reduce their Time to Market.
ASTM E2500(1) presents a number of similarities with the traditional V-model process, from the definition of needs through to maintenance of the validated status. The major innovation in this ASTM E2500 text lies in the fact that the tests to be carried out no longer follow a rigid FAT/SAT/IQ/OQ/PQ sequence as described in EU GMP Annex 15. All these tests, brought together under the single term “Verification”, can now be more freely and rationally organized to optimize efficiency and be adapted to each context.
The ASTM model
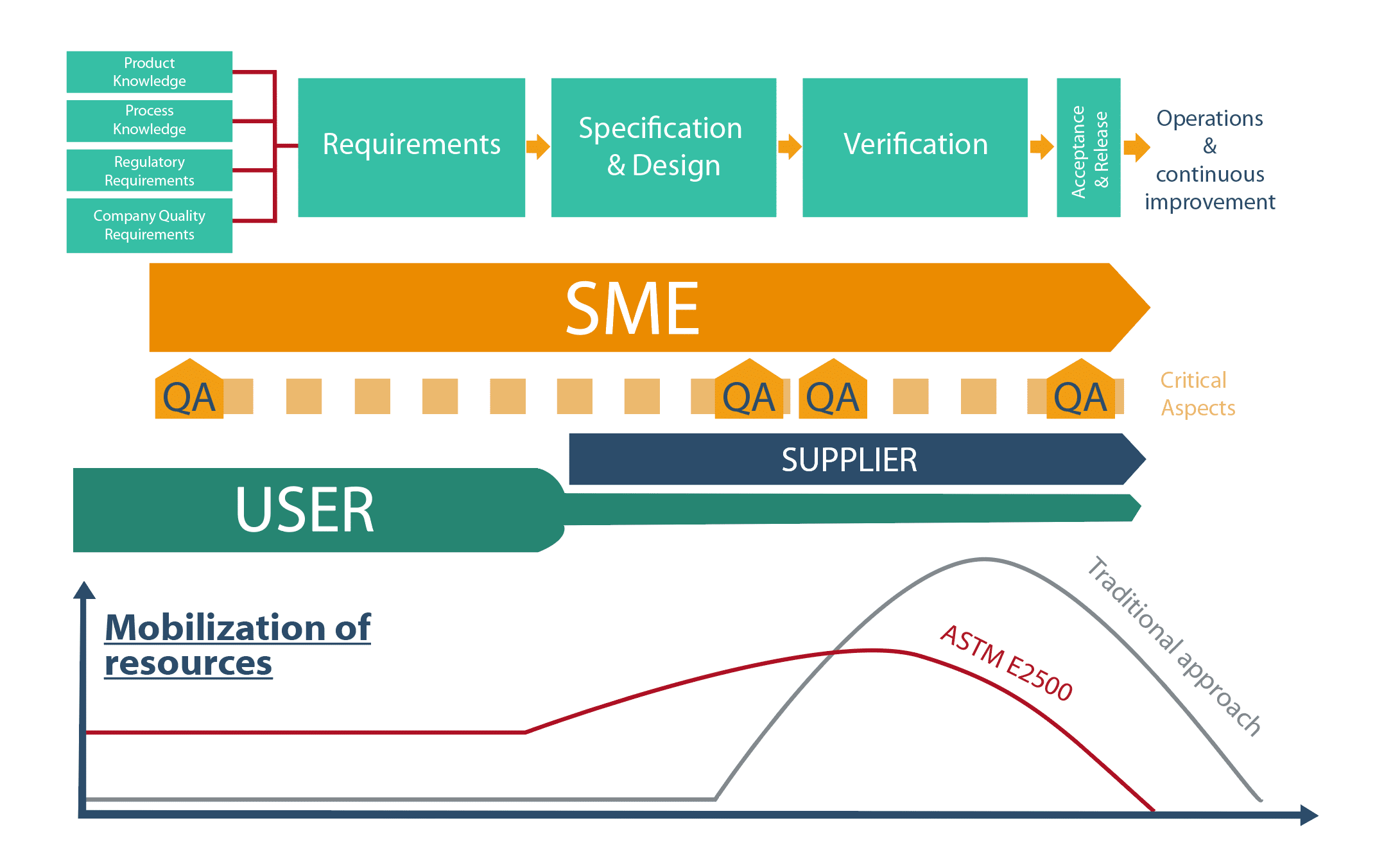
The ASTM E2500(1) model is hinged around 4 steps and 4 cross-functional processes:
- 4 steps : Requirements ; Specification & Design ; Verification ; Acceptance & Release.
- 4 cross-functional processes: Good Enginneering Practices ; Quality Risk Management, Design Review, Change Management.
The ASTM E2500 model highlights new players:Subject Matter Experts and suppliers.
The principle
The ASTM E2500 method is employed to control the implementation of a manufacturing system and secure projects by focusing on critical aspects from a GMP point of view at each step of the process.
The risk management approach focusing on product quality and patient safety makes it possible to adapt needs specification efforts and rationalize verifications.
The steps
Step 1: Requirements
Requirements underpin Verification; the documentary pyramid relating to verifications is built on these foundations. It is important that they are clearly defined. Input data include product knowledge, knowledge of the manufacturing process, regulatory requirements as well as the company’s in-house requirements. The involvement of users and SMEs in this step is vital. They will identify various user needs and facilitate the identification of critical aspects.
Step 2: Specification & Design
This step is aimed at verifying that the needs and requirements of the various users have been taken into account by the project unit and supplier. It is an iterative step; regular meetings are held to adjust solutions proposed by suppliers and ensure they are clearly understood. Where a prototype is concerned, it is evident that the design can only be partially defined. In fact, sometimes the verification step is needed to finalize it.
Step 3: Verification
The verification step is underpinned by two major principles: “the right test” carried out at the “right time” = “right first time”.
The application of these two principles enables a “right first time” test to be conducted. If carried out too soon the validity of its results may be undermined by modifications, dismantling or transport; carrying out tests too late undermines the project schedule.
Moreover, the Verification process consists in limiting test duplication. Tests are only repeated if necessary or in the event of a change. Accordingly, some tests may be conducted at the supplier’s site, either totally or partially. Similarly, some tests may be delegated to suppliers as they are experts in their own systems.
Step 4: Acceptance & Release
The Acceptance & Release step paves the way for the release of the system for operational implementation. During this step, it is important to draw up a summary document listing all tests carried out, the dates they were carried out and their status. The system concerned can then be transferred to the following step.
Should reservations remain at the point of transfer, they should only concern non-critical aspects and resolution dates must be defined.
Cross-functional processes
Process 1: Good Engineering Practicess
Good Engineering Practices are a matter of common sense: good, well-defined and shared organization from the start of a project facilitates its implementation. Poorly defined or undefined organizational problems at the start of a project will inevitably contaminate technical problems that will certainly arise.
Process 2: Quality Risk Management
A product-focused risk analysis is carried out to identify, at the earliest possible stage, critical parameters from a quality / patient safety point of view (CQA, CPP). The quality risk management approach also makes it possible to focus on the verification of critical aspects of the system / installation of interest from a GMP point of view.
Process 3 : Design Review
The design review is an iterative process. Accordingly, it has to be approached on a step by step basis as supplier design documents are published. This phase is critical to ensuring the system design meets needs. It is aimed at reducing the need for any modifications following handover of the system to end users, as well as avoiding repeat tests following a modification that can have a considerable impact on the launch of production. As above, it is evident that this process closely mirrors the Specification & Design step. Consequently, this activity should be repeated whenever there is a design change.
Process 4 : Change management
It is obvious that in the ASTM E2500 approach, the change management process will start earlier. Test duplication will be avoided provided change is managed, tests that may be conducted at the supplier’s site, even if only partially.
Conversely, since modifications are identified at an early stage via a well-prepared design, they should be easier to resolve as a result.
Players
The process set out in ASTM E2500 is based on an organization whereby R&R are clearly defined and better use is made ofvarious Project players’ expertise :
- SMEs play a crucial role in ASTM E2500(1). Essential from the start of a project, they contribute to the definition of needs, the identification of critical aspects, the review of system design, the drawing up of the verification strategy and the choice of acceptability criteria. They also take the lead role in the Quality Risk Analysis approach.
- Suppliers, veritable systems SMEs, may,depending on their maturity, be asked to conduct certain verification tests; this is aimed at achieving the “right test” executed “at the right time” (“right first time”) objective more easily. In projects conducted within the framework of ASTM E2500, suppliers play an essential role. They have to be familiar with the principles of ASTM E2500 and trained in regulatory requirements, particularly concerning good documentary practices.
- QA controls and focuses its efforts on critical aspects of the processes and systems that may have an impact on product quality. In particular, it validates all verification acceptance criteria related to critical aspects.
This organization is reflected in different resources being mobilized throughout the project. Such an approach makes it possible to take full advantage of the expertise of each individual player, and thus better safeguard projects.
Summary
ASTM E2500 is an approach that goes well beyond verification. It covers the entire project and involves all players from the very start of the project, from the needs definition phase. Driven by solid anticipation of the design step and the appropriate blend, the approach enables efficient verification via the execution of the right tests at the right time.
“Those who walk slowly can, if they follow the right path, go much further than those who run rapidly in the wrong direction.” René Descartes

Gérôme AVENOSO – LILLY France
Gérôme Avenoso started his nuclear career before joining Lilly in 1993 when he set up the C&Q structure. After his intervention on several local projects, he covers CQV activities on all corporate projects carried out in Europe. He then participated in a global program made up of 4 similar projects in Italy, the United States, France and China, the object being to respond to be more effective. Finally, it integrates a think tank on the C&Q method aimed at making the method simpler and faster in connection with the ASTM E2500 as well as promoting the harmonization of approaches across all Lilly sites.
avenoso_gerome@lilly.com

Jacqueline BESSET – ASSYSTEM
jbesset@assystem.com
Maëlle DESREUMAUX – ASSYSTEM
mdesreumaux@assystem.com
Stephane BERTRAND – LILLY France
bertrand_stephane@lilly.com
Share the article
Glossary
ASTM : American Society forTesting Material
C&Q : Commissioning & Qualification
CPP : Critical Process Parameter
CQA : Critical Quality Attribute
CQV : Commissioning Qualification Validation
FAT : Factory Acceptance Tests
GEP : Good Engineering Practices
GMP : Good Manufacturing Practices
IQ : Installation Qualification
OQ : Operational Qualification
PQ : Performance Qualification
QA : Quality Assurance
R&R : Role & Responsability
SAT : Site Acceptance Tests
SME : Subject Matter Expert
T T M: Time to Market
References
[1] ASTM E2500 : Standard Guide for Specification and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment
– ISPE Guide : Science and Risk-Based Approach for Delivery of Facilities, Systems, and Equipment
– ICH Guides ( International Conference on Harmonization) Annex 15 EU GMP
