


Juin 2012
La Vague n°34
Particules… des questions en suspension !
Sommaire
- Risques particules et verre dans les produits injectables.
- De la prévention à la détection des particules visibles.
- La maitrise de la contamination particulaire dans la production de bouchons en élastomère.
- Confluences particulaires, de la visibilité partagée.
- L’analyse de risque partagée entre fournisseurs et utilisateurs : une garantie de sécurité pour les patients.
- Revue des Pratiques d’Inspection Visuelle.
De la prévention à la détection des particules visibles
Une synergie dans l’analyse des risques selon le cycle de vie du produit injectable.
1. Contexte et historique
L’initiative proposée lors du 24e Congrès A3P d’octobre 2011 a permis de fédérer à ce jour près de 30 experts issus de 19 sociétés pharmaceutiques et partenaires fournisseurs.
Répartis en 4 sous-groupes (cf.§ 3.) qui ont débuté leurs travaux dès la fin 2011, le fil conducteur des travaux et des débats souvent animés et toujours précis suit les 4 piliers suivants :
- L’ICH Q8 Quality by design et les liens au développement des produits par la validation de leurs attributs qualité propres quant au critère particulaire,
- L’ICH Q9 Management du risque afin de bien différencier tout au long du cycle de vie du produit les impacts potentiels de toute défaillance du critère particulaire sur les procédés de fabrication, de contrôles, et la mise sur le marché,
- L’ensemble des Pharmacopées et Compendia définissant les caractéristiques particulaires,
- Les Bonnes Pratiques de Fabrication des produits injectables stériles régissant en termes d’objectifs autant les moyens de prévention que ceux de détection pour assurer la libération des lots en toute confiance.
2. Prévention versus détection
Tout au long du cycle de vie du produit, depuis la définition des composants, leurs contrôles, la réalisation des formules et des modes de répartition- scellage ou bouchage, l’inspection visuelle et les étapes de finition et de transports, chaque étape du processus de production au sens large se définit comme bornée par son fournisseur initial et son délivrable au client suivant (interne ou externe). C’est ce que nous avons voulu schématiser dans le diagramme où la voix du client (selon le principe d’analyse et de cartographie des processus) n’est pas la même (figure 1). Ainsi le risque pris en amont est-il un risque client alors que dans les étapes finales définitives ce risque est un risque fournisseur.
Attardons-nous sur ces deux niveaux de risques :
- On pourra prendre un risque client quand l’étape suivante peut réduire l’impact des modes de défaillances antérieurs. Typiquement, le risque client entraîne la probabilité de fournir au client un défaut au niveau de confiance statistique choisi. Dans le cadre des procédés en amont, il est probable de livrer des défauts au client interne (autre service, autre site) pour des étapes ultérieures si des solutions de remédiation existent (filtrer une solution est un moyen de séparation de particules, décanter, cribler, retourner des flacons en cas de risque de fragments de verre, ..)
- On ne dépassera pas un risque fournisseur quand le produit ne pourra plus être détectable ni modifiable sur sa composante particulaire et ce sera évidemment dès l’inspection visuelle, le conditionnement n’ayant plus une influence directe sur le taux de particules et la détection y restant faible voire inefficace. Typiquement, le risque fournisseur assure au client l’absence probable de défaut au niveau de confiance statistique choisi. Dans le cadre de l’inspection visuelle, il reste évidemment préférable, aux limites des contraintes et de performances industrielles, de garantir le rejet de bons produits plutôt que la livraison de probables défectueux.
La détection est par postulat un moyen a posteriori et la prévention restera le choix prioritaire dès les étapes de développement des formulations, de définition des cahiers des charges pour les matières premières, les contenants, les équipements en contact, les procédés de nettoyage, etc.
3. Analyse du risque particulaire
Chacun des 3 premiers sous-groupes a mis en œuvre une ou des méthodologies conseillées par l’ICH Q9 pour établir les facteurs de prévention et de détection communs et incontournables dans le traitement de la réduction des taux de particules dans les produits injectables, à savoir :
Le sous-groupe 1 : Explore les caractéristiques particulaires des composants et les engagements respectifs des fournisseurs et des laboratoires dans le développement et la maîtrise des produits selon ICH Q8. En ce sens, la prise en compte respective des besoins et des spécifications comme les études les tests discriminants1 à privilégier doit déterminer un haut niveau de qualité particulaire initiale. Les effets d’opérations ultérieures, du conditionnement, d’emballages et de transports seront évalués dans un second temps des travaux.
Le sous-groupe 2 : Évalue la prévention du risque particulaire tout au long des procédés de mise sous forme pharmaceutique et les impacts sur la conception, la construction et la maintenance des équipements critiques. Par l’AMDEC2 et des outils d’évaluation macroscopique, les points-clés de la prévention sont dégagés et priorisés. L’appel aux fabricants d’équipements au travers d’un questionnaire d’intérêt au problème particulaire devra aider chaque laboratoire-utilisateur dans une meilleure définition des besoins-utilisateurs3.
Le sous-groupe 3 : Situe les pratiques comparées d’inspection visuelle et définit la volonté d’une approche minimale commune dans la détection des particules visibles par les laboratoires du panel. Face aux pratiques différentes et justifiées par la nature des produits, des équipements, les historiques et les marchés, les travaux visent à définir des paramètres de procédé commun et des procédures dont le tronc commun couvrira l’ensemble des étapes et sous- étapes, depuis la définition de la criticité, les étapes de qualification des équipements, des défauthèques et des personnels et l’exploitation générale des résultats tant sur les rejets, les contrôles statistiques que les tendances. Un questionnaire sera adressé aux exploitants et en suite synthétisé. Une synthèse et des orientations communes sont attendues avec les commissions en cours de la SFSTP4 dans le domaine de l’inspection visuelle.
Le sous-groupe 4 : Nous fait partager sa vision du marché et la pertinence d’une analyse du risque patient à travers les réclamations et rappels pour présence anormale de particules visibles. Il s’agit de définir les critères pertinents tant toxicologiques que cliniques et bibliographiques pouvant autoriser la réduction et l’acceptation du risque particulaire. Les approches différentes des Autorités de Santé sont autant de facteurs à prendre en compte au moment où nos sites de production sont disséminés dans le monde et liés à des marchés multiples et encore peu alignés sur cette problématique, mais n’en doutons pas, en devenir de l’être.
La référence à ICH Q9 du point de vue méthodologique ne doit pas nous faire oublier que seule(s) l’étape ou les étapes en relation directe avec le patient et l’administration du produit sera concernée par une évaluation en risque fournisseur, ce que nous avons synthétisé dans ce diagramme (figure 1).
La somme des travaux du G.I.C aura comme délivrable un modèle de CHARTE QUALITE définissant les mesures et rationnels techniques et scientifiques validés par la Profession de l’Industrie Pharmaceutique afin de garantir une réduction du risque particulaire. Ainsi de cette charte pour démontrer la prévalence de la prévention sur la détection, la performance de la détection dans une approche commune minimale, la conjonction des actions d’amont en aval pour assurer la maîtrise de la teneur en particules visibles.
Ce thème fera l’objet d’un suivi permanent au travers de forums et de commissions techniques et de communications périodiques auxquels nous vous convions dès à présent.
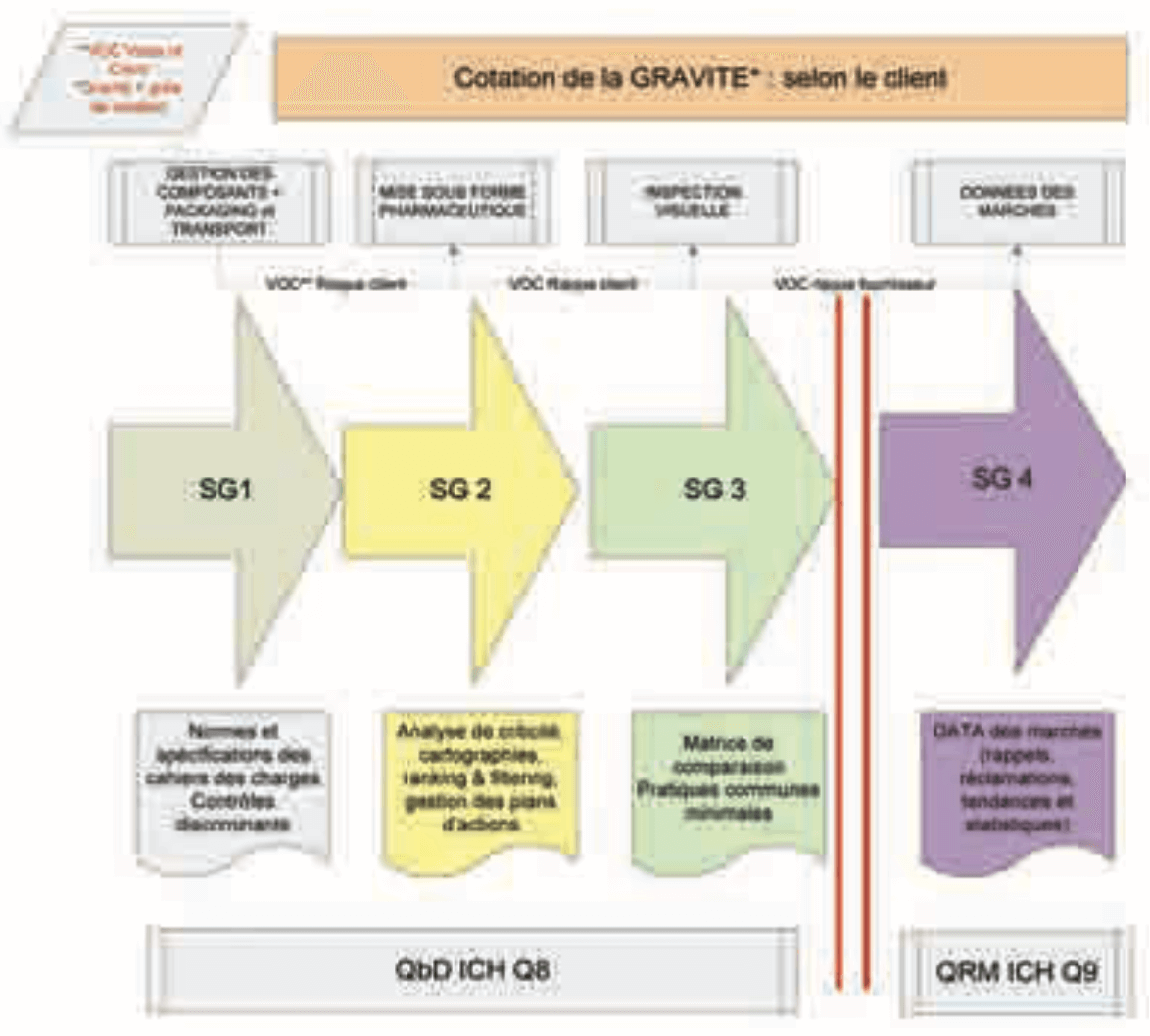
Figure 1
Partager l’article

Jacques NAVELLOU – AXYS NETWORK
Référence
[1] : On entend par test discriminant la méthode d’analyse qui va permettre de définir exactement et parfois même dans un contexte produit – dépendant, le niveau particulaire initial que ce soit sur la base d’une norme reconnue ou d’une spécification du client.
[2] : Analyse des Modes de Défaillances et Effets de leur Criticité (FMEA en anglais pour Failure Mode Effect Analysis).
[3] : SBU pour Spécifications Besoins Utilisateurs (ou en anglais URS pour User Specifications Requirements).
[4] : Société Française des Sciences et Techniques Pharmaceutiques.
