
Sommaire
- Considerations for Validating Aseptic Manufacturing Processes.
- Simulation des procédés de fabrication aseptique de suspensions de nanoparticules ou microsphères : Challenges & facteurs de succès au travers d’un retour d’expérience.
- L’isolateur jetable pour le remplissage aseptique, innovation ou hérésie ?
- A3P/AFI Survey on Sampling & Testing Practices for In-Process Pre-Filtration Bioburden for Sterile Products: Presentation of the Results & Critical Discussion.
- Projet de norme internationale amené à être amendé PR ISO 11737-3 : Stérilisation des produits de santé - Méthodes microbiologiques - Partie 3 : essai des endotoxines bactériennes.
- Real-time detection of CFU growth with the ScanStation® smart incubator expedites the environmental monitoring process.
- MTP: What It Is, Why It Matters. Module type package standard is a major step forward for plug-and-produce manufacturing.
- How Pemflow / D.O.C. manage Extractables and Leachables Qualification of the in-process materials and primary packaging.
Simulation des procédés de fabrication aseptique de suspensions de nanoparticules ou microsphères : Challenges & facteurs de succès au travers d’un retour d’expérience.
La plupart des produits injectables sont issus de procédés ayant une stérilisation terminale, post remplissage et scellage, soit par la chaleur (autoclavage) soit plus rarement par rayonnement (gamma ou beta). Ces procédés doivent être privilégiés au regard de la règlementation qui spécifie que tout procédé ne subissant pas une étape de stérilisation terminale doit être justifié. Cf BPF annexe 1, 94[1]

Cependant, tous les produits thermosensibles ou non radio-stérilisables devront être fabriqués dans des conditions aseptiques. Pour tous les médicaments filtrables, la stérilisation du produit sera réalisée par filtration au point le plus proche du remplissage donc par filtration en ligne pendant la répartition du produit. Ainsi la maitrise de l’asepsie concernera les étapes de la filtration stérilisante au scellage du contenant : filtration stérilisante, remplissage et scellage pour les ampoules ou bouchage pour les flacons ou seringues et lyophilisation si applicable.
Avec l’émergence des produits issus des biotechnologies ou des technologies telles que les microsphères ou nanoparticules, certains produits ne peuvent pas être stérilisés par filtration au point de remplissage. Ces nouvelles technologies font émerger de nouveaux défis auxquels les industriels doivent faire face tant sur le design du procédé que sur les approches d’assurance de la stérilité. En effet, dans ce cas, l’ensemble des étapes de fabrication de la substance active dans sa forme finale et du produit fini devront être réalisées en conditions aseptiques.
Les principaux challenges seront donc, de définir les conditions d’asepsie pour un procédé utilisant des technologies complexes, avec plusieurs étapes souvent peu automatisées et se déroulant sur plusieurs jours et de démontrer la maitrise de cette asepsie tout au long du procédé au travers des exercices de simulation ou APS (Aseptic Process Simulation).
Nous présenterons donc dans cet article, quels sont ces challenges et les facteurs de succès au travers d’une étude de cas.
1. APS ou MFT : aspects règlementaires et spécificités
En accord avec la règlementation et plus spécifiquement l’annexe 1 des GMP, une vérification périodique des procédés aseptiques doit être réalisée via un test de simulation utilisant un milieu de culture stérile. Auparavant, cette simulation, principalement réservée aux procédés de remplissage était plus connue sous le nom de “Media Fill Test”. Aujourd’hui de plus en plus de procédés aseptiques couvrent beaucoup plus d’étapes que celles de remplissage, bouchage et scellage. C’est pourquoi, on retrouve de plus en plus la dénomination APS pour Aseptique Process Simulation. Le draft de l’annexe 1 utilise notamment ce terme permettant ainsi d’étendre le scope de l’annexe 1 à tous les procédés aseptiques qu’ils soient simples ou complexes.
La production de principes actifs à libération prolongée utilisant la technologie des microsphères ou nanoparticules, requière un nombre important d’étapes de fabrication complexes en conditions aseptiques avant d’atteindre l’étape de remplissage et scellage tout en ayant la plupart du temps une étape de stockage de la substance active formulée de plusieurs semaines.
Même si le draft de la nouvelle annexe 1 prend en compte quelques spécificités, comme par exemple l’impact d’étapes pouvant impacter la viabilité de certains microorganismes: “§ 9.34 Where processing stages may indirectly impact the viability of any introduced microbial contamination, (e.g. sterile aseptically produced semi-solids, powders, solid materials, microspheres, liposomes and other formulations where product is cooled or heated or lyophilized), alternative procedures that represent the operations as closely as possible can be developed and justified.”, elle reste très orientée sur les étapes de remplissage.
En effet, comme on peut le voir dans les exemples ci-après, des règles sont données sur le nombre d’unités à remplir, sur la nécessité d’agiter les unités remplies pour assurer un contact du milieu de simulation avec la surface totale du contenant.
- § 9.42 The number of units processed (filled) for APS tests should be sufficient to effectively simulate all activities that are representative of the aseptic manufacturing process. Justification for the number of units to be filled should be clearly captured in the PQS. Typically, a minimum of 5000 to 10000 units are filled. For small batches (e.g. those under 5000 units), the number of containers for media fill should at least equal the size of the production batch.
- §9.43 Filled APS units should be agitated, swirled or inverted before incubation to ensure contact of the media with all interior surfaces in the container.
Face à une règlementation ne prenant pas complètement en compte ces spécificités et face à des procédés complexes et constitués d’une succession d’étapes de plusieurs jours, parfois manuelles, l’industriel doit s’appuyer sur une démarche structurée basée sur une évaluation des risques exhaustive et argumentée pour refléter au mieux les conditions de production pendant ces exercices de simulation afin de pouvoir assurer la stérilité du produit et sécuriser le patient.
2. Retour d’expérience
Une approche structurée sur le design et la réalisation des APS est clé pour garantir la représentativité du procédé et des opérations réalisées par les opérateurs. L’approche est décomposée en 5 parties que l’on se propose de décrire ici.
1. La description détaillée du procédé.
La description détaillée du procédé a pour objet comme son nom l’indique de recenser chaque étape du procédé et ensuite de détailler toutes les opérations rattachées à chaque étape de manière à pouvoir identifier, d’un point de vue assurance de stérilité et maitrise de l’asepsie, les opérations à risque et donc critiques. Une opération critique est ainsi définie comme une opération qui a une forte probabilité de rompre l’asepsie du procédé.
La difficulté principale dans cet exercice de recensement est d’établir une liste exhaustive des situations et des opérations. Plus le procédé est manuel et complexe, plus le recensement devra être précis et l’analyse de risque prépondérante. Afin d’y parvenir, le travail pluridisciplinaire est un atout majeur. Cet exercice devra intégrer à minima les opérateurs de production, la qualité opérationnelle, l’expert procédé, le responsable assurance de stérilité, un expert en microbiologie…
Dans le cadre de la fabrication de microsphères, par exemple, 9 étapes et 27 opérations ont été identifiées. Parmi ces étapes, on retrouve par exemple des étapes d’extraction de solvant, de concentration ou de lavage de microsphères. Alors que les opérations sont par exemple une connexion ou déconnexion aseptique, ouverture manuelle de vanne…
Pour chaque opération, la situation à risque, c’est-à-dire le risque de contaminer le produit, doit être décrite de manière précise afin de faciliter le travail sur les étapes suivantes. L’objectif est de déterminer les causes potentielles de cette situation à risque ainsi que les conséquences. S’il existe des moyens de préventions, ils seront indiqués dans le tableau afin de conclure sur l’acceptabilité ou non du risque.
2. Réalisation de l’analyse de risque
a. Choix de l’outil
La rédaction et le travail sur l’analyse de risque est le socle pour l’établissement de la stratégie et de la réalisation des APS. Ce travail est un travail d’équipe, basé sur des données scientifiques.
L’objectif de l’analyse de risque se doit d’être clair, de lister l’ensemble des actions pouvant être réalisés pendant un lot de production normale afin de couvrir le risque de contamination.
Une cotation des risques est alors effectué suite à son établissement. Cette criticité est un des facteurs justifiant la fréquence de réalisation de ces actions pendant les APS.
Le choix de la méthodologie pour la réalisation de l’analyse de risque impactera les données de sortie. Il n’existe pas d’outil universel, ils sont complémentaires et doivent être cohérents avec l’objectif de l’analyse. Parmi les différents outils, on distingue l’AMDEC, l’APR, le RRF, ou encore la HACCP.
Seuls l’AMDEC et l’APR seront discutés ici.
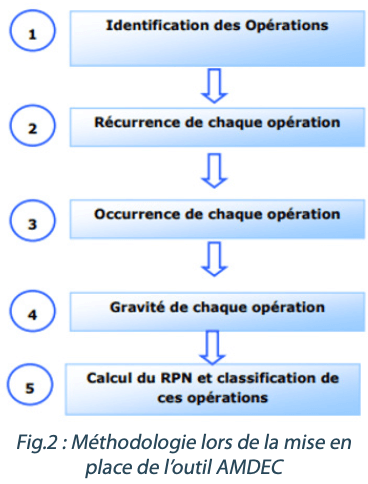
- L’AMDEC permet d’identifier des dysfonctionnements potentiels, d’analyser, de coter et d’évaluer des modes de défaillance, les hiérarchiser afin de les maitriser et peut se découper en 5 étapes distinctes, décrites dans le schéma FIg2.
- L’APR permet d’identifier et d’analyser des risques à partir d’une liste préétablie de situations dangereuses. Le but est d’identifier les risques associés à un processus.
Il peut se découper en 3 étapes distinctes : 1)Découper le processus en éléments unitaires ; 2)Créer la liste de situations de danger associées au processus d’audits ; 3) Sur une des étapes du procédé : identifier les risques, leurs causes et leurs conséquences.
Dans notre cas, le choix de l’outil s’est porté sur l’APR.
b. Réalisation de l’analyse de risque
L’objectif est de déterminer les causes potentielles de la situation à risques, ainsi que ses conséquences. S’il existe des moyens de préventions, ils seront indiqués afin de statuer sur l’acceptabilité ou non du risque.
Le challenge de cette partie consiste à décrire des scenarios s’étant déjà réalisé, mais également à penser à des situations n’ayant jamais eu lieu, mais qu’il serait possible d’observer.
Un des facteurs de réussite est l’implication des personnes expertes du procédé mais également de personnes candides afin d’apporter un œil neuf. Les opérateurs travaillant sur le procédé doivent être impliqués dans la réalisation de ce travail. La dimension opérationnelle prend alors tout son sens.
3. Plan de mitigation
Après avoir décrit les moyens de prévention / détection, l’acceptabilité du risque doit être statuée. Effectivement, la mise en place d’actions peut diminuer le risque de contamination tout au long du procédé. Ces actions de réduction des risques seront alors mises en place non seulement pour la réalisation des APS mais également en routine de production.
Sur la fabrication de microsphères, les systèmes à usage unique sont très utilisés et nécessitent de réaliser des beaucoup de connexions / déconnexions aseptiques pouvant impacter l’asepsie du procédé et provoquer la fin prématurée d’un lot. Il s’agit d’une situation à risque. Afin de réduire les risques, des modes opératoires détaillés sur la réalisation de ces opérations, associés à une formation complète des opérateurs, ont été mis en place. De plus, des contrôles d’intégrité des connections ont été implémentés. Comme cette opération est critique, elle sera simulée en APS et la réalisation de cette opération en production de routine est soumise à habilitation APS.
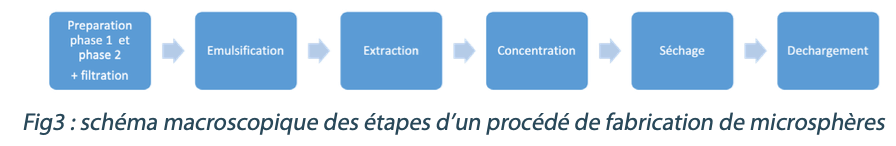
4. Simulation du procédé de fabrication
a. Spécificités du procédé de fabrication
Les conditions opératoires de réalisation du procédé peuvent impacter la stratégie de validation initiale et de routine des APS. Dans le cadre d’un procédé utilisant de l’azote comme gaz d’inertage, la qualification initiale du procédé aseptique est alors réalisée dans les deux conditions : en aérobie et en anaérobie et augmente donc le nombre d’APS à réaliser.
Dans le schéma Fig3, quelques étapes d’un procédé de fabrication de microsphères ont été représentées afin de montrer la complexité et la diversité des étapes concernées par la maitrise de l’asepsie et donc des APS.
Ce procédé de fabrication dure environ 100h du lundi matin au vendredi après-midi et comporte des étapes statiques et dynamiques. Chaque étape peut durer plusieurs heures et même plusieurs jours. Par exemple, la première étape de filtration est de l’ordre de plusieurs dizaines de minutes, l’étape d’émulsification est de l’ordre de quelques heures et celle du séchage de plusieurs dizaines d’heures. De plus,
- Pour les étapes statiques qui durent plusieurs heures ou jours comme par exemple une phase d’extraction, le milieu de culture est laissé dans la cuve pendant toute la durée de cette extraction avec ou sans la présence des opérateurs.
- Toutes les étapes dynamiques (exemple une phase de concentration), elles sont également simulées sur la durée complète.
Dans le but de couvrir de potentielles difficultés en cours de production, une majoration de temps réel de production ou des temps de pause sont appliqués à chaque étape.
b. Stratégie d’habilitation
La stratégie d’habilitation des opérateurs découle de l’analyse de risque.
- Durant les étapes de préparation des phases, ou d’émulsification, l’habilitation portera sur les nombreuses connexions / déconnexions aseptiques (une quinzaine de connexions et une trentaine de déconnexions). Chaque opérateur doit effectuer un nombre minimum de connexions et déconnexions à chaque APS afin d’être habilité.
- Durant l’étape de déchargement, des soudures sont effectués sur le sac de déchargement. Les soudures sont tracées et chaque opérateur doit réaliser à minima une soudure pendant l’APS
La difficulté réside lorsqu’il n’est pas possible de réaliser plusieurs fois la même opération durant l’APS. La génération des microsphères est une émulsification par le biais d’un système à membrane. L’opération très manuelle de ce montage ne peut être effectuée qu’une seule fois par APS. En validation de routine (2 APS par an), l’action ne peut être réalisée que par 2 opérateurs.
5. Interprétation et résultats APS : spécificités
a. Récupération des unités à incuber
A la fin d’une simulation aseptique, le milieu de culture est récolté
dans des poches ou contenant à usage unique qui sont directement incubées. Voir exemple de poches avec milieu de culture (Fig1).
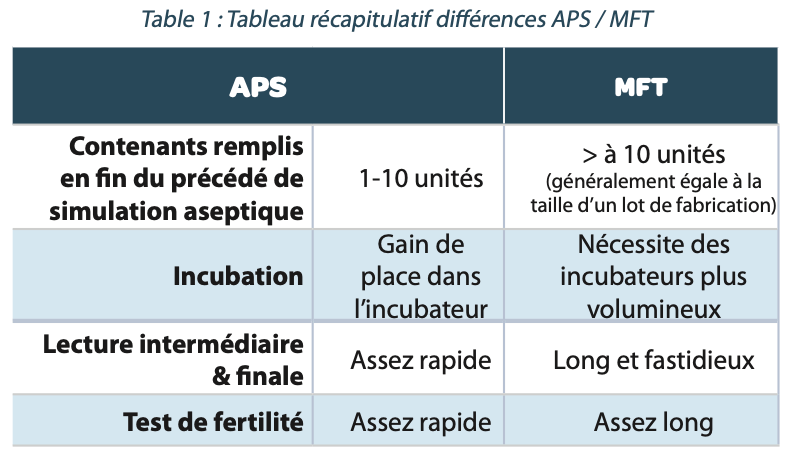
Ce tableau recapitule les différences en termes APS (bags) vs MFT (ampoules/flacons ..)
b. Incubation, lectures et formations
La cartographie des étuves utilisées dans le cadre d’incubation de poches de plusieurs dizaines de litres doit tenir compte de leur forme. La cartographie doit donc être faite en conséquence. En effet, la présence d’ampoules/flacons ou de poches impacte l’aéraulique au sein même de l’enceinte et donc l’état qualifié de ces enceintes.
La formation pour la lecture de ces poches peut cependant être plus complexe et ainsi nécessiter des formations/habilitations plus poussées. La manipulation des poches n’est pas aisée car elles peuvent être de dimensions importantes et impacter l’ergonomie du poste notamment lors du retournement des contenants remplis avant mise en incubation pour garantir la mise en contact du milieu avec toute la surface du contenant et du système de fermeture.
Enfin, le test de fertilité sera réalisé sur chaque élément incubé.
c. Réconciliation
La réconciliation des unités est plus facile que dans le cas d’un MFT et aucune unité contaminée n’est acceptée.
3. Conclusion
Au-delà des requis règlementaires, les industriels produisant des produits stériles issus de procédés aseptiques depuis la fabrication de la substance active formulée jusqu’à la répartition en contenant final scellé sont confrontés à de nombreux challenges. Ils doivent, dans un premier temps, définir un procédé suffisamment robuste d’un point de vue maitrise de l’asepsie en maximisant les systèmes clos par exemple. Ensuite les équipes opérationnelles doivent comprendre les enjeux et les risques associés au procédé et être formées et habilitées dans la durée. Les compétences techniques ne sont pas suffisantes, des opérateurs avec une gestuelle et une attitude adaptée sont clés. Enfin, le design des APS doit être représentatif afin de démontrer de manière fiable que le procédé dans sa globalité permet de produire un produit stérile pour sécuriser la santé du patient. L’APS ne doit être qu’une vérification de la stabilité du couple procédé/opérateur alors que le fondement de l’asepsie repose sur un design adapté et des opérateurs experts.
Même si l’approche et les principes de maitrise de l’asepsie sont identiques aux procédés de répartition, la technologie complexe des procédés de fabrication des substances actives aseptiques comme par exemple la production de microsphères ou nanoparticules, requière une équipe dédiée ayant une compréhension approfondie de ces procédés permettant d’assurer la qualité du produit et la sécurité du patient tout au long du cycle de vie du produit.
Partager l’article

Acronymes
AMDEC : Analyse des modes de défaillance, de leurs effets et de leur criticité
APR : Analyse Préliminaire des Risques
APS : Aseptic Process Simulation
EPPI : Eau pour Preparation Injectable
GMP : Good Manufacturing Practices
HACCP : Hazard Anaysis and Critical Control Points
MFT : Media Fill Test
RRF : Risk Ranking & Filtering
SU : Singe Use
