


Avril 2024
La Vague n°81
Externalisation des activités – Maitrise de la contamination
Sommaire
- Sous-traitance de la bioproduction des biomédicaments en France
- Externalisation des audits fournisseurs : Les clés de la réussite
- Selecting container closure components with confidence: A data-driven approach to CCI
- L’art de comprendre le langage : l’évolution du traitement automatique du langage
- Microbial Monitoring RABS Gloves: Unravelling the Implications of Directional Use
- General Considerations on Bacterial Endotoxins & USP Approach to Developing GC <86> Bacterial Endotoxins Test Using Recombinant Reagents
- Bacterial Spore Formers in Disinfectant Efficacy Testing
- Avoiding product oxidation by H2O2 in isolators. It all depends on the right analyses!
Sous-traitance de la bioproduction des biomédicaments en France
La bioproduction… un vaste sujet qui mérite de s’y pencher avec précision. Mise en lumière par la crise sanitaire liée à la pandémie du Covid-19, l’importance stratégique pour un pays de bénéficier de capacités de production sur son sol a été remise au centre du jeu par chaque pays du monde. Mais qu’entendons- nous par bioproduction ? Est-ce le même processus que de produire un anticorps thérapeutique, une thérapie cellulaire, une thérapie génique ou un vaccin. Malheureusement… non.

1. Commençons par le commencement
Pour conserver, voir reconquérir son indépendance sanitaire, il est indispensable de raisonner sur l’ensemble de la chaîne, depuis les phases de R&D d’un biomédicament jusqu’à sa bioproduction. En effet, un biomédicament est très souvent bioproduit dans le pays dans lequel il a été développé, dans la mesure où celui-ci dispose des capacités adéquates. La France possède deux atouts clés. Notre pays est en effet très performant, avec 898 projets de biomédicaments thérapeutiques en développement, fruits de la force historique et pérenne de notre filière académique. D’autre part, la filière française des sociétés de services innovants, permet de supporter les phases de développement d’un candidat médicament sur l’ensemble de la chaîne de valeur.
Nous disposons donc du terreau indispensable pour boucler cette chaîne avec des bioproducteurs capables de produire pour eux sur des sites propriétaires, mais également des sites “en propre”, en ce qui nous concerne ici, des CDMO (Contract Development and Manufacturing Organizations) capables de produire pour les autres en tant que prestataire de service !
Mais parler de bioproduction serait réducteur car il n’y a pas une bioproduction mais bien “des bioproductions”. En effet, produire un anticorps thérapeutique ciblant une large population dans des cuves de 20 000 L, avec un process de production globalement très cadré, n’a pas grand-chose à voir avec le fait de produire une thérapie cellulaire ou une thérapie génique ciblant une population le plus souvent restreinte. Choisir c’est renoncer comme on dit, et pourtant la France doit investir, avec ses partenaires européens, pour se doter de capacités sur chacun de ces différents biomédicaments aux processus de production très différents. Mais réjouissons-nous, l’Etat a pris conscience des enjeux et des AAP spécifiques ont été créés pour aider nos champions tricolores, qui jouent pleinement le jeu et investissent pour se développer et nous doter des capacités de bioproduction qui nous manquent.
Ainsi, depuis 2023, les CDMO et autres acteurs de la bioproduction ont annoncé plusieurs accroissements de capacités parmi lesquels : le LFB et Yposkesi qui doublent respectivement leurs capacités de production d’anticorps et de vecteurs viraux, GTP Bioways qui renforce ses capacités de production en système microbien, R&D Biotech qui a inauguré sa nouvelle unité de production de plasmides et Just Evotec qui, lui aussi, doit lancer prochainement son nouveau site de production. Ajoutez à cela des sociétés développant des innovations pour optimiser la bioproduction et le constat que la France veut reprendre la place qui était la sienne en matière de production de médicaments ne fait plus de doute. Mais si les besoins sont importants et que le France veut se positionner, la concurrence est belle et bien présente, et l’Asie arrive en force depuis quelques années.
Samsung Biologics ou WuXi Biologics pour ne citer qu’eux gagnent inlassablement des parts de marché et font souffrir bon nombre de CDMO françaises et européennes (le Sénat Américain a d’ailleurs récemment pris des mesures visant notamment Wuxi, afin de limiter la dépendance Américaine à ce nouvel acteur fort). Les rachats et fusions en cours en sont une preuve flagrante. La filière doit s’organiser, se structurer tout en restant agile afin d’absorber les baisses de volumes quand les temps sont durs, et de prendre en compte les spécificités toujours plus complexes des biomédicaments à produire. Elle doit également investir sur l’avenir en créant des plateformes en capacité de développer les processus souhaités par leurs clients.
2. État des lieux de la filière française des CDMO
L’article L5121-1 CSP de la législation professionnelle de l’industrie du médicament (cadre juridique de l’Union Européen) définit un biomédicament comme “tout médicament dont la substance active est produite à partir d’une source biologique ou en est extraite et dont la caractérisation et la détermination de la qualité
nécessitent une combinaison d’essais physiques, chimiques et biologiques ainsi que la connaissance de son procédé de fabrication et de son contrôle’” Les biomédicaments englobent ainsi une gamme diverse de substances, comme des protéines humaines modifiées, des anticorps monoclonaux, des facteurs de croissance, des vaccins, des enzymes, des organelles, des virus et des cellules ou tissus vivants. Etant donné la complexité intrinsèque de toute source biologique qu’elle soit cellulaire ou acellulaire, la (bio)production des biomédicaments nécessite un environnement hautement contrôlé, une stricte conformité réglementaire et des procédures de contrôle de la qualité lourdes, afin de garantir l’innocuité et l’efficacité du produit final.
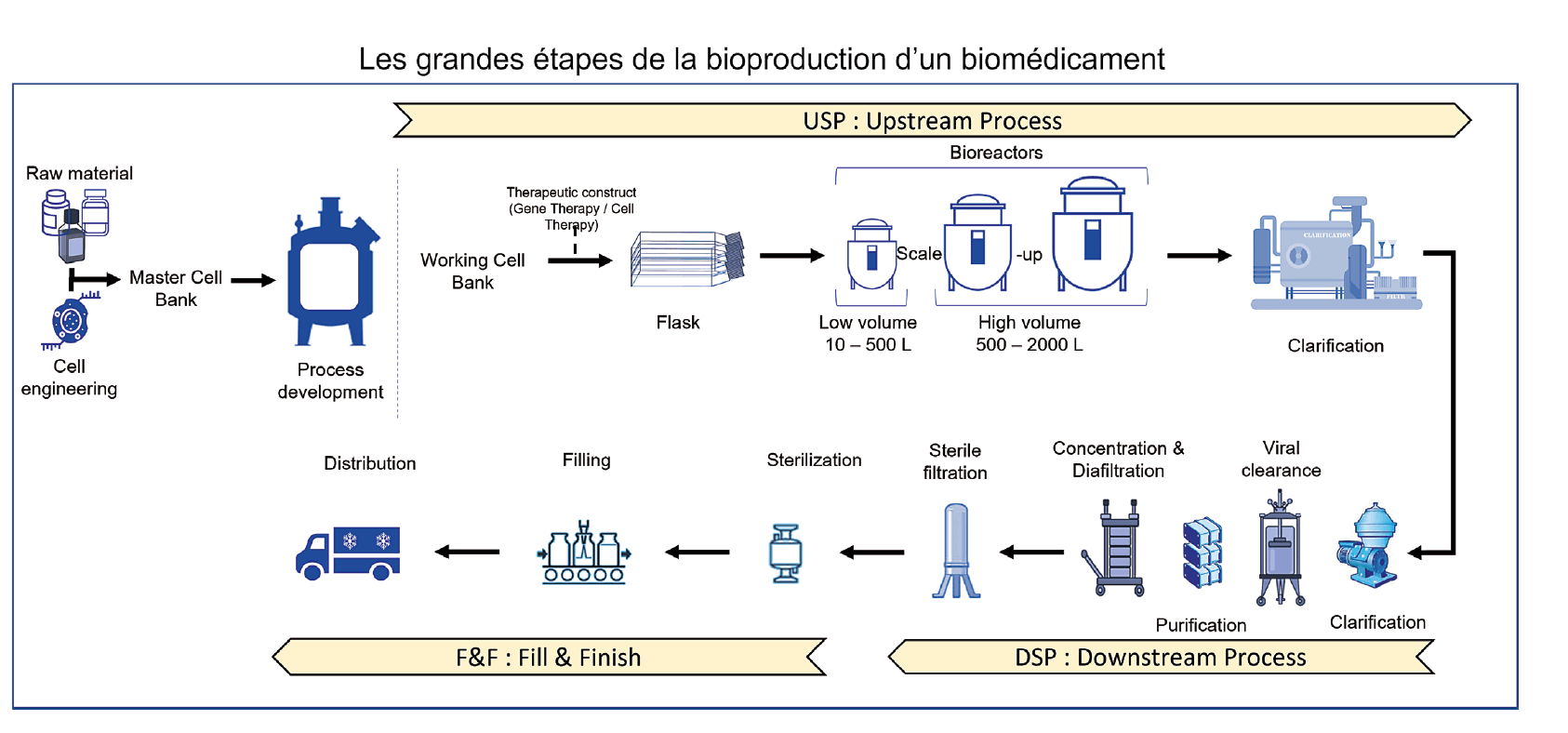
La bioproduction des biomédicaments peut être divisée en 3 grandes étapes clés : UPS, DSP et F&F.
USP (ou Upstream Process)
L’objectif lors de l’USP est d’optimiser la croissance de la source biologique et la production du biomédicament. Cette étape implique la préparation et la culture du matériel biologique, tel que des cellules de mammifères ou des microorganismes, nécessaires à la production du biomédicament. Elle comprend des étapes comme la culture cellulaire, la fermentation et le développement d’inoculum. Des stades supplémentaires de génie génétique impliquant des modifications du génome sont également positionnés en USP. La culture cellulaire ou microbienne se fait en volume croissant (scale-up) dans des bioréacteurs à usage unique (single-use) ou à multi-usage (multi-use). Plusieurs modes de culture sont maintenant utilisés en bioproduction avec une culture à volume constant sans perfusion (Batch culture), une culture avec ajout de milieu (Feed Batch Culture) et une culture à volume constant avec perfusion en continue (Continuous culture). Indépendamment du mode de culture, le contrôle et la régulation des différents paramètres critiques du milieu sont essentiels. En fin d’étape, le milieu sera saturé avec le produit d’intérêt, mais également avec des débris et des déchets de culture.
DSP (ou Down Stream Process)
L’objectif en DSP est d’une part d’éliminer les impuretés issues de la culture en bioréacteur et d’autre part de concentrer le produit biomédicament d’intérêt. Le traitement post-USP consiste à séparer et à purifier le produit du “bouillon” de fermentation ou de la culture cellulaire. La récolte (Harvest) va permettre la séparation des cellules ou du micro-organisme du milieu de culture, afin d’obtenir uniquement le surnageant ou la biomasse. Par la suite, un enchaînement de filtrations, de centrifugations, de chromatographies et d’autres techniques de séparation vont permettre d’isoler le produit et éliminer les impuretés. En ce qui concerne la filtration, deux techniques sont utilisées pour purifier et concentrer les produits. La filtration à flux direct (Direct Flow Filtration) qui implique le passage direct du liquide à travers un filtre, retenant les impuretés tandis que le produit désiré est collecté. La filtration à flux tangentiel (Tangential Flow Filtration), quant à elle, fait circuler le liquide tangentiellement à la surface d’une membrane, permettant au produit de passer à travers tout en retenant les impuretés. Les étapes de chromatographie sont spécifiques à chaque type de biomédicament.
En fin de DSP, des stades supplémentaires de modification du produit peuvent également avoir lieu comme la bio-conjugaison des anticorps monoclonaux pour donner des immuno-conjugués.
F&F (ou Fill & Finish)
À cette étape, le produit purifié est formulé sous sa forme posologique finale, qui peut inclure des formulations liquides ou des poudres lyophilisées. Le produit peut également être conditionné selon son mode d’administration dans des flacons, des poches de perfusion ou des seringues. De plus, toutes les étapes finales de traitement nécessaires, telles que la stérilisation ou l’emballage, sont réalisées pour préparer le produit à la distribution et à l’utilisation.
3. Critères de choix d’une CDMO
Les sociétés type CDMO jouent un rôle crucial dans l’industrie pharmaceutique en fournissant une large gamme de services aux entreprises biotechnologiques et pharmaceutiques. Elles sont apparues pour répondre aux besoins croissants des développeurs de biomédicaments en expertise spécialisée, flexibilité opérationnelle, réduction des coûts, accélération du développement et gestion des risques.
Le choix d’un partenaire CDMO est décisif dans le développement d’un projet et peut grandement en impacter le succès ou l’échec. Le processus de sélection est donc complexe, long et dépendant de la stratégie de la société qui développe le biomédicament (mise sur le marché, licensing, vente…). Étant donné l’ampleur du processus de sélection d’une CDMO, nous suggérons de résumer les critères principaux généralement utilisés pour ce choix.
Les principaux critères de choix sont donc :
Expertise & Expérience
Les CDMO sont uniques avec des forces et des spécialités différentes, se distinguant principalement par leur expertise technique et leur expérience pratique. Les critères de sélection incluent l’expérience GxP pour répondre aux exigences réglementaires, la familiarité avec les types cellulaires requis, l’accès aux matériaux cellulaires appropriés, les outils de biologie cellulaire, moléculaire, biochimique pour assurer le développement et la production.
Qualité & Conformité
La qualité est fondamentale dans la fabrication pharmaceutique. Une CDMO doit respecter les normes de contrôle qualité strictes, comme les BPF, et avoir des certifications telles que ISO, FDA, EMA. Il est important d’évaluer leurs systèmes qualité, leur historique d’inspections réglementaires, et leurs capacités analytiques et de transfert de technologie. Les études de stabilité et la conformité aux BPF sont également des facteurs déterminants pour garantir le succès du projet. Votre partenaire de fabrication doit avoir de l’expérience dans la préparation des dossiers IND ou disposer d’un soutien juridique. La CDMO doit proposer un soutien pour naviguer dans des paysages réglementaires complexes, avec une expertise étendue en matière de conformité réglementaire.
Relation client
Une communication efficace est nécessaire pour établir une relation de confiance et favoriser la collaboration et la transparence tout au long du processus de développement d’un médicament. Il est important d’évaluer l’approche en gestion de projet de la CDMO, notamment la désignation de chefs de projet dédiés, la définition des délais et des jalons. De plus, il est essentiel de comprendre leurs protocoles de communication, la fréquence des rapports et l’accès aux mises à jour en temps réel. Il convient également de vérifier si leur style de communication est en adéquation avec vos attentes et s’ils sont en mesure de fournir des mises à jour transparentes, opportunes et proactives sur l’avancement du projet.
Capacité de production
Lors du choix d’une CDMO, il est crucial de considérer leur capacité à augmenter la production à mesure que votre projet progresse dans les différentes étapes de développement et potentiellement de commercialisation. Évaluez la capacité de leurs installations, leurs ressources disponibles et leur flexibilité pour répondre à ces besoins croissants pour éviter les goulots d’étranglement.
Flexibilité & Adaptabilité
Pour soutenir efficacement les programmes clients à long terme sans compromettre la qualité, la flexibilité doit être intégrée dans le processus opérationnel et la culture de la CDMO. Il est essentiel de choisir un partenaire réactif et agile, capable de gérer des programmes cliniques complexes tout en ayant les capacités d’expansion nécessaires.
Rentabilité
Choisir la CDMO le moins cher peut sembler attrayant, mais une analyse détaillée est nécessaire pour éviter des problèmes futurs. Externaliser permet de réduire les frais généraux et d’allouer les ressources de manière plus efficace, favorisant une planification financière plus prévisible. Cependant, il est crucial de mener une due-diligence approfondie avant de sélectionner un partenaire CDMO pour éviter les retards et les coûts supplémentaires. Prioriser les économies de coûts au niveau des tâches peut parfois compromettre la qualité et le soutien technique de la CDMO, entraînant des retards et des coûts supplémentaires à long terme. Il est donc indispensable de considérer l’ensemble des facteurs lors de la sélection d’une CDMO pour éviter les retards et les risques.
4. Caractéristiques spécifiques de la bioproduction selon le type de biomédicament
L’hétérogénéité, la complexité et la maturité variable des biomédicaments font que les enjeux et les forces de la chaîne de valeur de leur bioproduction respective diffèrent selon le type de protéine thérapeutique, le vecteur viral, le type de cellule ou les populations de cellules à produire.
L’anticorps thérapeutique a pour avantage d’avoir maintenant fait ses preuves… Depuis le premier anticorps commercialisé en 1985 (muromonab), pas moins de 170 anticorps sont arrivés sur le marché pour apporter des solutions époustouflantes sur bon nombre de pathologies alors en échecs thérapeutiques. Visant souvent des larges populations en oncologie ou en inflammation (ex. le pembrolizumab qui est en essai clinique dans 1407 essais en parallèle), le processus de production est maintenant connu et maîtrisé dans sa globalité. S’il existe certes toujours des axes d’amélioration pour produire avec un meilleur rendement et pour moins cher, le coût d’une poche d’anticorps se compte en quelques milliers d’euros… Ayant montré leur efficacité, certains anticorps arrivant en fin de protection, ont été “copié” et des biosimilaires sont arrivés sur le marché avec des prix pouvant tomber à quelques centaines voire quelques dizaines d’euros. Le prix de ces biosimilaires démontre à lui seul que le coût de production, avec un effet volume certain, peut être maîtrisé et maintenu à un niveau acceptable pour les systèmes de santés.
La thérapie génique offre des avantages majeurs, notamment le traitement direct des maladies génétiques et une correction durable des gènes défectueux. Elle ouvre également la voie à de nouvelles approches thérapeutiques et à des traitements pour un large éventail de maladies. La thérapie cellulaire permet, quant à elle, un traitement précis et ciblé, la capacité de régénération tissulaire, une toxicité réduite, et des perspectives de traitement pour des maladies jusqu’ici incurables. Elle permet également une personnalisation des traitements, des effets durables, et une réduction de la charge thérapeutique. Cependant la fabrication à grande échelle de ces thérapies doit prendre en compte l’hétérogénéité des types de vecteurs et des types de cellules à produire.
En l’absence de normes établies , de gold standard, dans la bioproduction de ce type de thérapie, il est devenu d’usage de dire que le process ( de production) est le produit (thérapie) les deux étant tellement indissociable et spécifique.
De plus, les capacités de production de certaines cellules thérapeutiques restent limitées compliquant la montée en échelle et altérant la reproductibilité du process. En ce qui concerne les vecteurs viraux, leur rendement de production reste à améliorer. A ces verrous technologiques, il faut également ajouter une réglementation lourde avec des contrôles de qualité stricts. L’utilisation de produits autologues dont le matériel de départ est prélevé du patient pour être ensuite administré à ce même patient post-bioproduction, vient ajouter une complexité logistique supplémentaire à la chaîne de valeur. L’ensemble de ces éléments impacte significativement le coût de production des thérapies cellulaires et géniques et autres médicaments de thérapies innovantes (MTI). Et c’est ce coût de production élevé qui va en majeure partie, et dans la plupart des cas, impacter le prix du produit thérapeutique.
Aux Etats-Unis, le prix d’un traitement autologue à base CAR-T, thérapie à base de cellules modifiées génétiquement et utilisées exclusivement en oncologie coûte aux alentours de 400,000$ alors que le seul traitement de thérapie génique pour l’hémophilie B culmine actuellement à 3.5 millions de dollars. Du fait d’une politique d’accès aux marchés différente entre les Etats-Unis et l’Europe, certains produits de thérapie cellulaire ou génique ne sont toujours pas disponibles en France ou ont été enlevés du marché. L’avènement des thérapies allogéniques dont le matériel de départ provient de donneurs sains, des solutions d’automatisation et des nouvelles technologies innovantes en DSP et USP devront permettre d’améliorer la bioproduction de ces thérapies.
5. Deals récents dans le domaine de la bioproduction
Le dynamisme du domaine de la bioproduction se traduit notamment à travers des opérations financières récentes de fusion et d’acquisition chiffrées à plusieurs millions d’euros voire plusieurs milliards d’euros. En effet, début 2023, Sartorius, par le biais de sa filiale française Sartorius Stedim Biotech, a conclu un accord pour acquérir la société strasbourgeoise Polyplus proposant entre autres des solutions de transfection, pour environ 2,4 milliards d’euros. Polyplus avaient eux- mêmes acquis la CDMO française Bio-Elpida et la CRO belge Xpress Biologics l’année précédente. L’autre acquisition notable dans le paysage français sur cette même période, est celle d’ABL Europe, une CDMO française et filiale de l’Institut Mérieux, par le britannique Oxford Biomedica pour 15 millions d’euros qui devra permettre à ces- derniers d’élargir leurs capacités de production en vecteurs viraux. Plus récemment, c’est le géant Danois de l’investissement Novo Holdings qui contrôle également Novo Nordisk, a qui annoncé en début de 2024, l’acquisition de Catalent, l’un des plus grands CDMO américains, pour 16,5 milliards de dollars. Novo Holdings prévoit de finaliser l’acquisition d’ici la fin de 2024 pour ensuite céder au moins 3 sites de production à Novo Nordisk pour 11 milliards de dollars avec comme objectif de booster la production de son nouveau blockbooster, le Wegovy. En ce début d’année, deux autres CDMO ont annoncé leur rapprochement. Le Finlandais Biovian s’est associé à l’espagnol 3P Biopharmaceuticals, pour former 3PBIOVIAN. Cette fusion stratégique vise à offrir des services de développement et de fabrication ‘End to End’ pour les systèmes d’expression protéique et les vecteurs viraux, couvrant les phases précliniques à commerciales.
6. Conclusion
En tenant compte de la diversité du type biomédicament qui peut être produit en France, du nombre de sites de production et du nombre croissant de deals financiers, nous assistons à une véritable révolution au sein de la filière nationale de la bioproduction. Cette expansion promet non seulement un impact positif sur l’économie nationale, mais également sur la santé publique et la souveraineté sanitaire.
Partager l’article




