


Sommaire
- Biotherapy. Médicaments de Thérapie Innovante et Annexe 1 : Analyse de son applicabilité
- How Can the Industry Drive Down the Cost of Goods to Better Serve the Patients?
- Maintaining contamination control in advanced therapy medicinal product manufacturing
- A Plug-and-Produce GMP Plant for Cell and Gene Therapy
- Maîtrise des risques : Stratégies et innovations pour sécuriser le développement des biomédicaments
- The Challenges of Floor Cleaning & Sanitization
- Statistical Approach to Aseptic Process Simulation: Representativeness and Proactive Alert Limit Setting for Aseptic Interventions
- Comparative Study of WFI Pretreatment Performance Electrically Based Pretreatment Outperforms Media-Based Pretreatment
- Détection à 100% des défauts critiques PP
- L’industrie pharma doit réduire sa trace carbone, ... le traitement d’air. Part 1
Maintaining contamination control in advanced therapy medicinal product manufacturing
In a fast-paced, rapidly evolving environment like Advanced Therapy Medicinal Product (ATMP) manufacturing, it is easy to overlook critical elements necessary to maintain contamination control. These critical and time-sensitive manufacturing procedures require a sound strategy for contamination control to help ensure the microbiological safety of the final product, and the protection of both patients and manufacturers.

This paper addresses the process and regulatory guidelines unique to ATMP and makes recommendations on cleaning and disinfection strategies including special considerations not found in other medicinal product manufacturing facilities.
1. Cell And Gene Therapy Process
ATMP’s are a broad category that includes cell therapy and gene therapy manufacturing. For these sites, patient-derived cells are isolated and then manipulated to create or enhance therapeutic attributes that can result in extraordinary clinical outcomes. This process can be allogeneic or autologous, but both involve bringing cells of human origin, therefore contaminated, into a controlled environment, performing manipulations that may be highly manual in nature and then rapidly delivering the therapy back for injection into the patient or patients. While many of these manipulations are performed in an isolator, restricted access barrier system (RABS) or a biological safety cabinet (BSC), a holistic contamination control strategy is still essential. RABS and isolators are designed to eliminate the potential for entry of microorganisms and must be guarded from contamination sources. In more open areas, such as ISO-7 or 8 clean rooms with a BSC, aseptic manipulation and aseptic techniques are critical to limit the ingress of contamination into the aseptic production area. Regulatory guidelines, including the EudraLex Volume 4 Annex I, outline a holistic approach as well as common sense disinfection practices to ensure that classified areas are under control and contaminants are limited.
2. Regulatory
The cell and gene therapy industry is a growing and expanding market. These manufacturing environments operate under aseptic conditions and need to follow the applicable cGMP regulations. The main regulations in Europe that apply to cell and gene therapy are EudraLex Volume 4 and Annex 2A(1,7). In the United States the cell and gene therapy markets are regulated by the Center for Biologics Evaluation and Research (CBER), which regulates cellular therapy products, human gene therapy products, and certain devices related to cell and gene therapy.(1,2,3) CBER uses both the Public Health Service Act and the Federal Food Drug and Cosmetic Act as enabling statutes for oversight. The cell and gene therapy suites operate as aseptic processing facilities and follow cGMPs. The Code of Federal Regulations and the FDA’s Aseptic Processing Guide would also apply to aseptic processing in cell and gene therapy facilities.(4,5) Looking into the future, the cell and gene therapy facilities that are located in Europe or that sell products into Europe may also be required to develop a detailed CCS and follow Annex I. Cell and gene therapy facilities will also need to follow ISO-14644-5:2024 which covers the concept of impact assessments. Both impact assessments and CCS will become a common part of cleanroom management.(6,7) The CCS is a holistic approach to contamination control with a focused objective on limiting contamination in the aseptic areas and the surrounding cleanrooms. Bioburden, particles, and endotoxins would all be reduced and controlled with a CCS program that involves ongoing continuous improvement.
3. Special Considerations In Atmp
ATMP facilities can differ from traditional pharmaceutical or biopharmaceutical facilities in several key ways. The rapid turnaround time and streamlined processes often result in a different set of utilities and facility features that can result in special considerations for ensuring a contamination-free environment. ISO 5 aseptic processing areas require the use of sterile items, sterile disinfectants and sterile sporicides. An ATMP facility may not have access to an autoclave to sterilize non-sterile items coming into the graded area. In this case, it is important to confirm that sterile items are available to purchase and that sterile disinfectants can be obtained. Frequently, the availability of in-house water-for-injection (WFI) is also limited within growing ATMP facilities. This can create some challenges for disinfectant preparation and rinsing of surfaces such as floors and walls. Processes to sterilize water or the variability in pre-packaged sterile WFI can also influence the presence of organic or inorganic material. The creation of a disinfectant use-dilution requires the highest-grade water available to prevent the presence of any organic or inorganic materials that might impact the efficacy of the disinfectant. Fortunately, there are many highly effective disinfectants available in a ready to use format that should be considered.
All one-step cleaner/disinfectants will contain surfactants and active ingredients that may need to be periodically rinsed from surfaces. Although the residue is not inherently problematic, a rinsing program should be implemented on a periodic basis using the same approach (i.e., a 2 or 3 bucket system) as the disinfectant application. This important step can appear more challenging with the absence of high-quality water production onsite, but sterile WFI can be purchased and used for a scheduled (e.g. monthly or quarterly) rinsing application. ATMP facilities frequently employ multiple suites to manufacture critical autologous medicines. The size of these suites can impact disinfection by creating challenges with dedicated bucket systems, bucket change frequency and removal of unused disinfectant. Sterile bucket liners are an excellent tool for simplifying the need to clean and re-use buckets. Having barrier products such as covers and wraps for items to be stored or to protect the sterility of items introduced into the isolator or RABS is essential. A two or three bucket system, with disinfectant in two buckets, should be changed at a frequency of roughly 1000 ft2 (93 m2 ) for ISO-7 and ISO-8 cleanrooms (600 ft2 (56 m2 )in ISO-6, and ISO 5 cleanrooms).(8) The smaller size of some ATMP suites may result in left over material that should not be moved to another controlled area. The use of disposable bag liners can aid with removal of extra disinfectant solution. After the cleaning and disinfection step, automated biodecontamination methods such as vaporized hydrogen peroxide (VHP) provide an additional level of contamination control for facilities that have multiple patient product suites. VHP can provide an entire suite biodecontamination that reaches all surfaces including HVAC systems and HEPA filters. VHP does not require rinsing after the process and is best deployed between product and/or patient batches that require an enhanced level of bioburden control. Fully integrated, mobile, and service options are available for VHP biodecontamination.
4. Disinfection
There is no one size fits all approach to disinfection as the selection and frequency of use of a rotational program will depend on environmental conditions and monitoring data. When first implementing a cleaning and disinfection program, the following guidance can aid in establishing an initial program for this critical process of ensuring that critical environments are free of contamination. Multiple products and product configurations can be employed to handle the various features and characteristics of a typical ATMP suite. (Figure2)
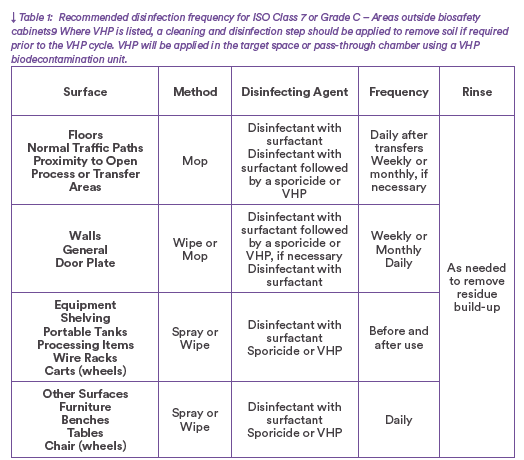
Floors, walls, and ceilings in ATMP
A good rotational program should consist of one effective, broad-spectrum disinfectant and one sporicide(4,7,10,11). Many ATMP facilities include an ISO 5 area for processing, either in a BSC, Isolator or RABS, within an ISO 7 suite. A proposed frequency of decontamination for the suite is listed in the Table 1.
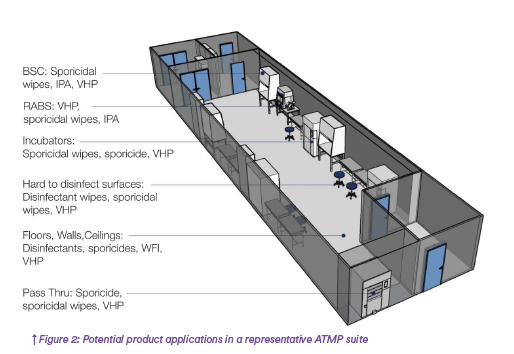
Biological Safety Cabinets in ATMP
BSC’s can be critical in the preparation of ATMP products and require extremely low bioburden levels in the ISO 5 zone. To achieve this, a sporicide must be employed before and after key activities. Sporicidal wipes are a highly effective means to achieve the required contamination control. VHP can also be deployed between patient batches to biodecontaminate the entire BSC. This is achieved by running the BSC blower during an automated room VHP process or isolating the BSC with an independent VHP treatment.
Isolators and RABS in ATMP
Since the current version of Annex I has been published the trend in the industry is to remove the operator as a possible source of bioburden in the cleanroom. Isolators and closed RABS are becoming more common in the cleanroom industry and in cell and gene therapy spaces. Production and cell manipulation that takes place in BSC hoods can be moved into Isolators and RABS and reduce the overall risk of contamination from operators and the ISO 7 and 8 cleanroom areas. Gloveless isolators and robotics may become more prevalent in the cleanroom industry and can be a useful production environment for cell and gene therapies.
Per Annex 1, the isolator biodecontamination process should be automated, validated, and controlled within defined cycle parameters using a gaseous or vaporized sporicidal agent(7). For example, VHP is a common method used in isolator biodecontamination. A cleaning process using a sterile cleaning agent is required prior to VHP treatment if any soils, spills, or debris are present. RABS also require a validated and robust method of disinfection using a sporicidal agent(7). This is achieved using an appropriate sterile liquid cleaner/disinfectant and sporicide. VHP treatment of a RABS simultaneously with the room is also possible as part of an automated biodecontamination cycle. By removing the operators from the cell and gene therapy production area it will significantly lower the overall risk of contamination from particles, endotoxins, and bioburden. These closed manufacturing areas also employ automated gaseous biodeocontamination methods providing less operator intervention and a more repeatable process. Reducing the overall risk of contamination is attainable with a robust CCS and utilizing in depth impact assessments.
5. Conclusion
It is possible to have a relatively simple program that consists of a single broad-spectrum disinfectant for use on the floors with a rotational pairing of a sporicide and rinsing. Although the frequency of application will be dependent on each individual facility, an approach of daily disinfection of the floors with disinfectant, monthly sporicide and water rinsing, can be effective. The size of ATMP processing suites, up to 1000 ft2 (93m2 ) often makes it easy to utilize a dedicated 2 or 3 bucket system to an individual suite and not require replacing the disinfectant in the buckets with a fresh solution during use, as may be necessary in larger areas. Sporicidal wipes are available and can be liberally applied for decontaminating BSC’s, pass through items and hard to disinfect surfaces. Finally, VHP can be deployed as an automated process for routine material transfer and for room, BSC, RABS and isolator biodecontamination between patient batches or production campaigns. Overall product selection and application methods should take into consideration the unique challenges of the ATMP facility including materials transfer, items and equipment within the facility, regulatory requirements, and other site-specific details.
Maintaining robust and ongoing control of the cell and gene therapy cleanroom is achievable with a scientific, risk-based CCS. Given the special considerations that are inherent with ATMP manufacturing, proper planning and attention to risk factors in advance of implementation of a disinfection program is critical for maintaining contamination control.

References
1. annex 2a: manufacture of advanced therapy medicinal 3 products for human use: version 15 (pe 009-15)
2. pic/s guide to good manufacturing practice for medicinal products annexes, pe 009-17 (annexes), 25 august 2023.
3. EudraLex, The Rules Governing Medicinal Products in the European Union. Volume 4: Good Manufacturing Practice Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products
4. Sterile Drugs Produced by Aseptic Processing: Current Good Manufacturing Practice, FDA October 2004.
5. Code of Federal Regulations, Part 211: Current good manufacturing practice for finished pharmaceuticals.
6. iso-14644-5: cleanrooms and associated controlled environments, 2024.
7. annex i: the rules governing Medicinal Products in the European Union. Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Manufacture of Sterile Medicinal Products. August 2022.
8. Dixon, A.M. “Cleaning of Non-Product Contact Surfaces”. Cleaning and Cleaning Validation. Edited by Pluta, DHI Publishing, LLC.
9. STERIS Corp. Technical Tip 4014
10. USP 43 <1072> Disinfectants and Antiseptics
11. PDA Technical Report #70 Fundamental of Cleaning and Disinfection Programs for Aseptic Manufacturing Facilities
Partager l’article


