


Sommaire
- Assurance Qualité & Production : impacts de la nouvelle annexe 1 des BPF
- Un expert en inspection visuelle doit-il être un expert technique en système de vision ?
- Taux résiduel de peroxyde d'hydrogène présent dans les isolateurs lors de tests de stérilité. Quel impact sur les données générées ?
- Médicaments de thérapie innovante à base de bactéries.
- Supplier & End-User Disinfectant Qualification Comparison for Cleanrooms
- Achat de remplisseuses BFS pour un site spécialisé dans le PFS.
- Mise en œuvre d’un procédé de fabrication d’ingénierie tissulaire dans le cadre d’un MTI expérimental en thérapie cellulaire.
Supplier & End-User Disinfectant Qualification Comparison for Cleanrooms
Disinfectants are a critical element in cleanroom contamination control and must be well-suited for the applications for which they are intended. Disinfectants should be tested both during development and registration to ensure proper selection and use. Subsequently, they should be tested by the end-user to ensure that the disinfectant is appropriate for the intended purpose within a specific facility.

Testing a disinfecting agent during product development can differ greatly from the testing required by the end-user to validate a finished formula. Different challenges exist for both developmental testing and effectiveness validation testing that must be understood. This idea is true for all types of disinfecting agents, including those defined as sanitizers, general disinfectants, sporicides, and sterilants (1). In this article, we compare the methods utilized by a disinfecting agent manufacturer and a pharmaceutical manufacturer to characterize these disinfecting agents and outline some of their respective challenges to help understand expectations and avoid pitfalls. Although sanitizers or cleaners such as sodium hypochlorite, hydrogen peroxide, and alcohol are often just active ingredients and water, disinfectants are typically complex and complete formulations. Developing a disinfectant has many challenges. There are multiple considerations with cleaning ability, broad-spectrum efficacy, stability, safety, toxicity, aesthetics, compatibility and multiple other desirable characteristics that must be addressed before a product is released to the market. Adequate efficacy results is one of the most critical and time-consuming elements of the disinfectant development process. Disinfectants must meet strict regulatory requirements for efficacy and require data to be submitted and approved before releasing with claims.
1. Regulations & Testing
Supplier development testing
Disinfecting agent manufacturers must register their disinfecting agent based upon a specific wet contact time, microbial kill, and the disinfecting agent concentration in compliance with the relevant authority’s regulations (2-4). During the development of a disinfectant, testing typically starts with active ingredient evaluations.Active ingredient selection is critical, yet options are limited by availability and acceptability in different world regions based on toxicity and environmental impact, among other factors. Disinfectant active ingredients, unlike antibiotics, are in their mode of action, and there must be a balance between safety for both the end user and environment and efficacy. Studies such as a time-kill kinetic study are often utilized for early product development to rapidly screen active ingredients and very basic formulations.
The time-kill method is a basic evaluation of disinfectant activity(5, 6). Suppliers may utilize a time kill early in the product development process to screen multiple active ingredients against problematic microorganisms. Although the time-kill method is a very useful tool for antimicrobial testing, often the test underestimates the complex applications faced by disinfectants and can be too little of a challenge.The time-kill method is simply a suspension test that utilizes a set volume of disinfectant that is directly inoculated with a challenge microorganism, with surviving colonies enumerated after established contact times.While easy to perform, unless testing bacterial or fungal spores, the disinfectant will often exhibit complete kill in this method at the shortest feasible contact times, such as 15 seconds.
More useful for supplier product development studies are methods that utilize a hard surface on which the challenge microorganism is dried before disinfectant application. These tests include established consensus methods that utilize stainless steel or glass coupons but can also include modifications of the methods required for product registration to add a quantitative endpoint(5,7). For these tests, a step to quantitate surviving microorganisms using a chemical neutralizer, sonication, or vortexing and plating can provide additional data for regulatory methods that are qualitative in nature.
It is best to take a complete and varied approach to select test methods and challenge microorganisms during development.The ASSOCIATION OF OFFICIAL ANALYTICAL COLLABORATION (AOAC) INTERNATIONAL methods required in the United States utilize a qualitative endpoint with no growth as the measurable data for whether a product passes or fails. This type of method is of little value when determining the role of different formulation excipients or when comparing the efficacy of different prototypes. The European quantitative methods (EN Norms) used for registration provide more valuable data when evaluating candidate disinfectants during development. However, they may not correlate to meeting the pass/fail criteria in the United States. During disinfectant product development, modifications or alternative methods must be considered to create a formula that can meet all applicable standards for efficacy, allowing for registration and use in multiple countries and municipalities.Ultimately, after development testing has finalized or nearly finalized a formula that looks promising as a potential marketed disinfectant, well-controlled and strictly regulated efficacy testing must be performed as necessary for markets in which the product may be sold.
In the United States, disinfectants are considered pesticides and are regulated by the Environmental Protection Agency (EPA) under the Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA). The EPA has well-defined and long-established criteria for proving safety and effectiveness, including prescribed test methods and strict data considerations (28). There are many approved and standardized methods that disinfectants are required to be tested against for achieving specific product claims to be denoted on a label. For everything from carpet sanitizers to residual efficacy and biofilm claims, the total number of prescribed tests is too many to list here. Below are the main methods utilized for hard surface disinfectant and sporicidal claims. These methods also have a complementary approach based on dosage form with wipe and spray products requiring different methodology with different experimental endpoints.
https://www.a3p.org/wp-content/uploads/2022/10/Cleanrooms_vague75_tab1-1.png
Many of these methods have been in use for decades and have a long history and extensive data for multiple classes and types of disinfectants. This data is very valuable for a regulatory agency when evaluating new products and comparing performance between new and older products to ensure safe and effective use. However, many of these methods predate advancements made in microbiology method development and can be limited in their scope outside of base claim generation. For example, most of the EPA methods are qualitative in nature and provide only a pass or a fail as an experimental endpoint. Although helpful to determine whether a formula is effective or not, the qualitative methods offer little other information and are challenging to use for comparisons or relative evaluations. Further, the methods present a very high challenge to ensure effectiveness in multiple application areas, from industrial to hospital environments. They may be too great of a challenge for what might be encountered in a cleanroom or controlled environment. A starting titer, well over one million microorganisms is not representative of a cleanroom or controlled environment’s bioburden levels.
In Europe, the registration process falls under the Registration, Evaluation, Authorisation, and Restriction of Chemicals (REACH) guidelines which also require demonstration that the products are safe and efficacious (8). Like the US EPA and corresponding AOAC and ASTM methods, the European Norms also have standard methodologies established for disinfectants and require multiple phases and steps based on labeling and product usage. The EU methods include both basic suspension-based studies for initial evaluation of disinfectant activity via a step one approach and then quantitative hard surface methods for specific applications for step two, phase two in the registration process. Because these methods are newer and provide log10 reduction values versus simply a pass/fail result, they are more easily adaptable to end-user applications. Still, do not perfectly mimic the real-world conditions or challenges met in a cleanroom or controlled environment (6-8).
For the rest of the world, the test methods and regulations can differ by region. Regulations may differ in the rest of the world and may conform in part, in whole, or not at all with US and EU guidelines. However, the desire for disinfectants that work as intended, with minimal safety concerns, and well-documented efficacy claims, remains the same. The testing requirements and methods are similar, however, and have the same limitations regarding real-world applicability whether they are more qualitative or quantitative in nature.
Additional considerations must be evaluated to provide labeling and use instructions incorporated into the required test methods. These include the use of organic soil during testing, hard water for disinfectant preparation, and the establishment of contact time and temperature. Developing disinfectants that can meet strict regulatory requirements is a challenge found worldwide. One of the most challenging aspects is consistently and reproducibly passing the various efficacy standards that are required by the different regulatory authorities. Although the methods can have a long history of use, with demonstrable success to ensure safe and effective formulations, their use does not always translate to other applications. Often, the methods are focused on reference strains and surfaces, require extremely high microbial challenges beyond what is found in the pharmaceutical environment, and can have pass/fail endpoints that do not apply to real-world situations.
End-user qualification testing
The regulatory guidance entails (bio)pharmaceutical manufacturers to qualify the disinfecting agents’ wet contact time on specific surfaces and microorganisms(9-11). Norms and industry technical documents share guidance for in vitro and in situ testing(12-16), as seen in Figure 1.
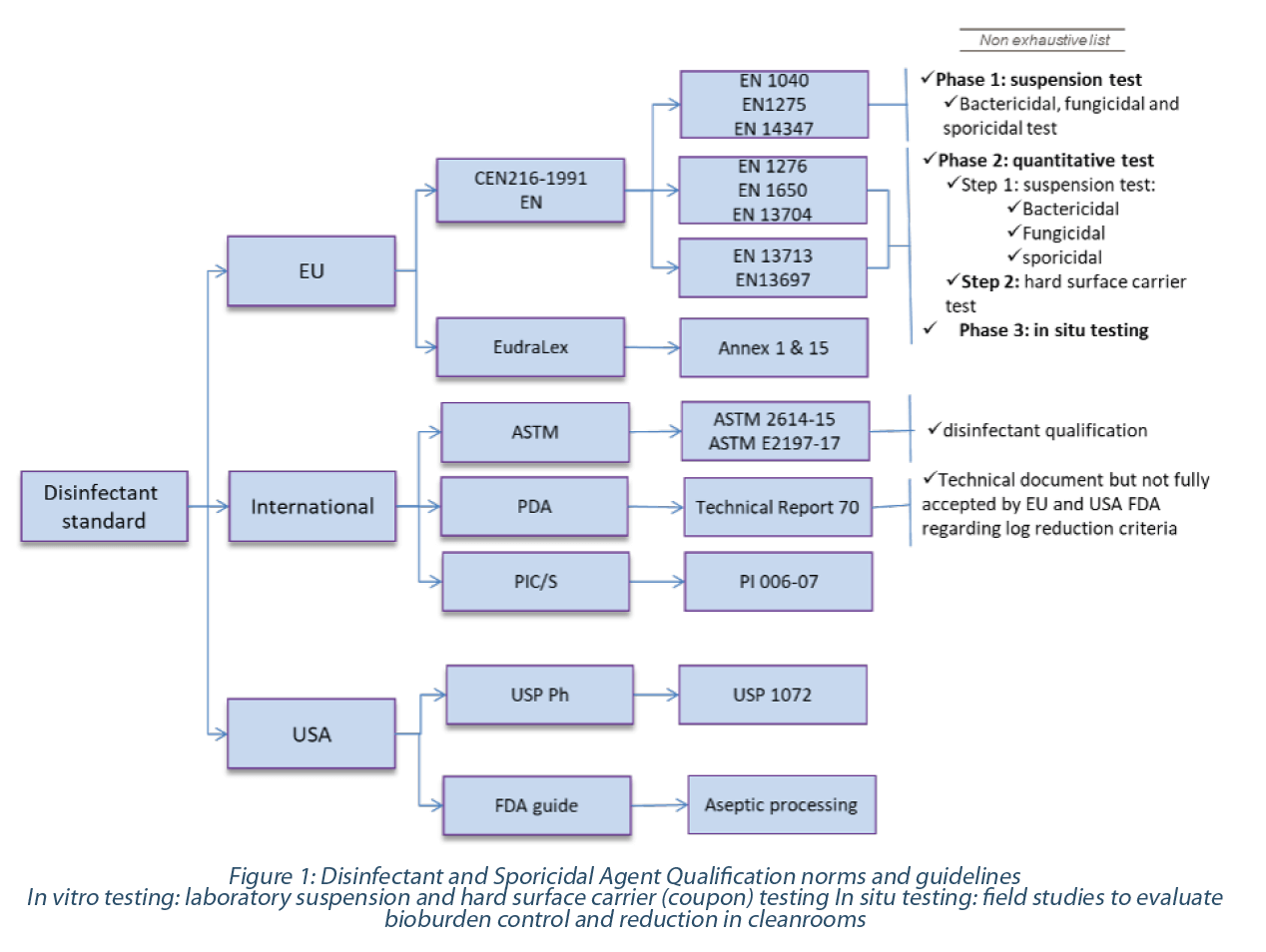
2. End-user disinfecting agent in vitro qualification
The pharmaceutical manufacturer is responsible for evaluating the optimal wet contact time, use dilution, use dilution expiration, based on the supplier’s data, their cleanroom surfaces, and most frequent isolate(s) from their historical Environment Monitoring (EM) data (2, 5-7, 9-15). The suspension qualification demonstrates the efficacy of the disinfecting agent to inactivate a planktonic microorganism at a determined contact time. The disinfecting agent efficacy from the suspension test may differ when a microorganism is adsorbed on a surface. Therefore, coupons that are representative of surfaces in the cleanrooms should be tested in vitro.
In vitro qualification of a disinfecting agent is based on the (2):
- wet contact time,
- log reduction acceptance criteria,
- microorganism type,
- disinfectant concentration,
- water quality,
- clean or soiled surfaces,
- surface types,
- Temperature of application.
The in vitro testing will confirm the efficacy of the disinfecting agent against a microorganism type adsorbed on a specific surface at a defined wet contact time. In-situ testing (real-world) is essential to assess the impact of the element listed below on the disinfecting agent efficacy (2):
- Temperature and humidity in the cleanroom: that may interact with the disinfecting agent kinetics of kill
- Air exchange in the cleanroom: that may impact the time of the disinfecting agent to dry
- Application technique (e.g., vaporization, spraying, mopping, wiping): that impact the removal of residue or microorganism on the surface
- Surface cleanliness: that may interact with the disinfecting agent’s efficacy.
3. Disinfectant Efficacy Testing
Efficacy is demonstrated by testing the disinfectant efficiency in reducing the microbial bioburden in either suspension (planktonic state) or on cleanroom surfaces (sessile) to an acceptable level.
The points to consider when developing a disinfectant efficacy test (DET) are:
- Log reduction: A disinfectant efficacy study is built on a microorganism logarithmic reduction that a disinfecting agent can achieve. A different set of logarithmic reduction is proposed for in vitro disinfecting agent qualification. (3)

There is no compendial or harmonized regulatory requirement on the logarithmic reduction required for pharmaceutical manufacturers(15). Therefore, pharmaceutical manufacturers should define the most appropriate log reduction based on their activities and historical EM data analysis. In many cases, the USP <1072> log reduction criteria are most suitable for the pharmaceutical industry. The EN documents are subject to various industries (food, industrial, domestic, and institutional areas), including pharmaceutical operations. Therefore, it is logical to observe that pharmaceutical manufacturers use a different type or combination of standards (Figure 2).
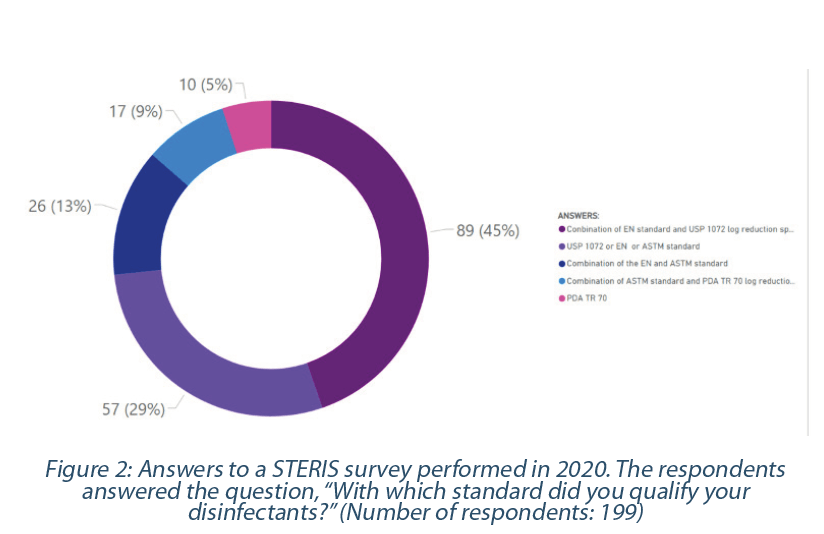
ATCC microorganism: A disinfecting agent should be qualified against ATCC microorganisms(4). Such qualification may be transposable from site to site or from supplier testing if the coupons tested to represent the cleanroom’s surfaces (15).
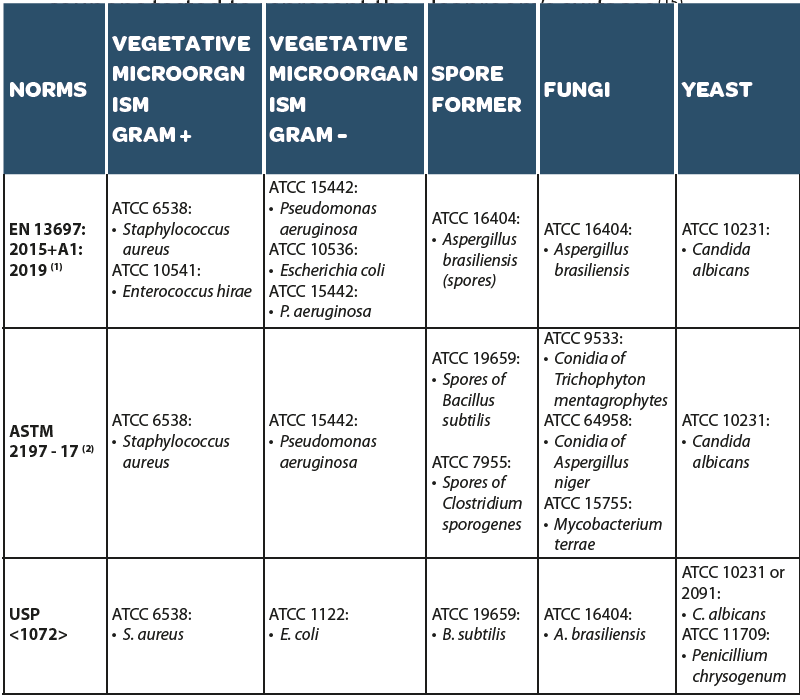 | (1) for specific application addition strains may be chosen if required: Salmonella typhimurium ATCC 13 311; Lactobacillus brevis DSM 6 235; Enterobacter cloacae DSM 6 234; Saccharomyces cerevisiae (for breweries) or ATCC 9 763 or DSM 1 333; Saccharomyces cerevisiae var. diastaticus (for breweries) DSM 70 487. (2) note that ASTM E2197 list virus as organisms that could be tested depending on the applications. |
- Isolate microorganisms (in-house microorganisms): To adequately evaluate the performance of a disinfectant in a real-world environment, it is essential to consider environmental isolates from the controlled environment in which the product is used. These isolates should be collected from environmental monitoring and test cultures selected based on the frequency of isolation, location, and general microbiological characteristics (4-15). For a broad-spectrum evaluation of in-house microorganisms, representative microorganisms from different classes, i.e., Gram-positive, Gram-negative, fungal or bacterial spores, may need to be considered.
- Disinfectant concentration: The definition of the optimal concentration of use should be designated by the supplier. It is advised that the end-user comply with the recommended supplier concentration. The end-user may elect to evaluate a range of concentrations and their efficacy against a set of microorganisms.
- Water quality: during the qualification of the disinfectants pharmaceutical manufacturers may elect to choose the lowest water quality used for the disinfectant dilution. It is considered good practice to use purified water or water for injection for disinfectant dilution.
- Clean or soiled situation: the draft guidance on the BPR supports that cleanroom end-users not including a soil load “As an exception to the rule, products to be used in cleanrooms do not require additional soiling in the test. A cleanroom has a controlled level of contamination that is specified by the number of particles per cubic meter at a specified particle size. The soiling level in cleanrooms is so low that even testing under clean conditions for the EN tests is still overdosing on soiling compared to cleanrooms. For these uses the high load of test organisms can be seen as soiling. Tests without soiling will only be accepted when the label states the specific use in clean rooms which are classified according to ISO 14644-1 in class 1 to 9 or according to GMP EU classification in Grade A to D. Generally, soiling will reduce the efficacy of the disinfectant, and where soiling is present, longer contact times, higher concentrations, pre-cleaning or a combination of these elements may be necessary.”(18). A visual inspection of the cleanroom surfaces should justify either a ‘clean’ or ‘dirty’ condition to test(16).
- Surface types and selection: The disinfectant qualification involves in vitro coupon (small part of surfaces) testing. The material of construction and porosity of the surface will affect the efficacy of the disinfectant by influencing the wetness of the microorganism to be killed. The coupons are spiked with a known amount of microorganism aliquot, and the disinfectant is then left for a predefined wet contact time. Then, the disinfectant is neutralized, and the log reduction is calculated.
It can be challenging to choose the representative and correct surfaces to test. There is no harmonized approach between pharmaceutical manufacturers; some may select the surfaces based on:
1.The most common surfaces in the cleanrooms.
2.A grouping matrix that includes surfaces of similar porosity and material of construction.
3.An assessment (Figure 3) that takes into consideration several elements, high probability to the parameters indicates that the surface should be tested:
a) the surfaces prevalent in the cleanrooms. The surfaces tested should be in good condition; therefore, surfaces with poor conditions must not be integrated into the testing(15).
b) increase in surface porosity and roughness can be a challenge in disinfection efficacy.
c) cleanroom classification where the surface sits. It is critical to ensure that disinfection of surfaces in critical classification (Class 100 and 1,000) are optimal to prevent contamination.
d) surfaces that are often touched by the manufacturing personnel when activities or interventions are frequent.
- Temperature of application: various standards mandate at least to test the disinfecting agent at a temperature within 15° to 25°C(7-27). The efficacy of a disinfecting agent at a defined concentration may increase with elevated temperature and vice versa(2). Pharmaceutical manufacturers should explore testing their disinfecting agent at different temperatures when most cleanrooms are maintained at temperatures below ambient.
4. Requirements and minimal information in protocol
The goal of in vitro efficacy testing is to demonstrate that the chosen chemistries are effective against environmental isolates on representative cleanroom surfaces, using test conditions that mimic standard operating procedures (water quality, presence of organic soil load, and use-dilution hold times– Figure 4).

5. Conclusion
Disinfectants are a critical element in cleanroom contamination control and must be well-suited for the applications for which they are intended.
Testing a disinfecting agent during product development can differ greatly from the testing required to validate a finished formula by the end user. The testing performed by the disinfecting agent manufacturer is to register the disinfecting agent and claims based upon a specific wet contact time, microbial kill, and the disinfecting agent concentration. In contrast, end users achieve disinfectant testing in vitro and in situ to demonstrate bioburden control in pharmaceutical cleanrooms. Therefore, the testing conditions may differ from the one used by the supplier or the end user.
Partager l’article



References
- Willison-Parry, D., Yang, S., Forng, R.-Y., Cirbo, T., Mcmeel, A., Kiler, B., & Phillion, C. (2019). Disinfectant Efficacy: Understanding the Expectations and How to Design Effective Studies That Include Leveraging Multi-Site Data to Drive an Efficient Program. PDA Journal of Pharmaceutical Science and Technology, 74(2), 249–263.
- El Azab, W., and Shields D, A Refresher on Disinfectant Wet Contact Time, A3P La Vague (https://www.a3p.org/disinfectant-wet-contact-time/)
- West, A. M., Teska, P. J., & Oliver, H. F. (2019). There is no additional bactericidal efficacy of Environmental Protection Agency–registered disinfectant towelettes after surface drying or beyond label contact time. American Journal of Infection Control, 47(1), 27–32.
- Brown, E., Dhanireddy, K., Teska, P., Eifert, J., Williams, R. C., & Boyer, R. (2019). Influence of drying time on prewetted disinfectant towelettes to disinfect glass surfaces. American Journal of Infection Control, 47, 846-848.
- ASTM International E 2197-17e1, Standard Quantitative Disk Carrier Test Method for Determining Bactericidal, Virucidal, Fungicidal, Mycobactericidal, and Sporicidal Activities of Chemicals, (2017).
- European committee for standardization, European standard EN 1276, chemical disinfectants and antiseptics. Quantitative suspension test for the evaluation of bactericidal activity of chemical disinfectants and antiseptics used in food, industrial, domestic and institutional areas. Test method and requirements (phase 2, step 1), (2019).
- European committee for standardization, European standard EN 13697, Chemical disinfectants and antiseptics – Quantitative non-porous surface test for the evaluation of bactericidal and/or fungicidal activity of chemical disinfectants used in food, industrial, domestic and institutional areas – Test method and requirements without mechanical action (phase 2, step 2), April (2015).
- Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC, accessed on 25 Aug 2021: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32006R1907
- EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 1, Manufacture of Sterile Medicinal Products, 2008
- EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines, Annex 1 Manufacture of Sterile Medicinal Products, Draft 2020 V12
- DA, Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice, 2004
- PI 007_6: “validation of aseptic processes”
- United States Pharmacopeia (USP) 42 <1072> Disinfectants and Antiseptics, 2019
- Parenteral Drug Association Technical Report 70 Fundamentals of Cleaning and Disinfection Programs for Aseptic Manufacturing Facilities, 2015
- Environmental Protection Agency, Pesticides: science and policy: efficacy data requirements supplemental recommendations, access on October 18,2018: https://archive.epa.gov/pesticides/oppad001/web/html/dis-02.html
- Derek Willison-Parry, Stephen Yang, Ren-Yo Forng, et al. Disinfectant Efficacy: Understanding the Expectations and How to Design Effective Studies That Include Leveraging Multi-Site Data to Drive an Efficient Program, PDA J Pharm Sci and Tech 2020, 74 249-263
- El Azab, W., A justified process for cleaning and disinfection; cleanroom Technology, March 2019, access on January 21, 2011: https://cleanroomtechnology.com/news/article_page/A_justified_process_for_cleaning_and_disinfection/152716
- Crystal M. Booth, Environmental Isolates: What’s The Proper Use Of In-House Cultures? Pharmaceutical Online, June 29. Access on January 21, 2021: https://www.pharmaceuticalonline.com/doc/environmental-isolates-what-s-the-proper-use-of-in-house-cultures-0001
- DRAFT Guidance on the BPR: Volume II part B+C (version 3.0 July 2017). Accessed on Jan 2022: https://www.echa.europa.eu/documents/10162/23047722/bpr_vol_ii_parts_b_c_rev_pt5_peg_en.pdf/fd98286d-b7a8-4b27-3506-a5061f107bf8
- El Azab, W., Residue removal in cleanrooms: A regulatory overview, Jan 2020, Access on June 25, 2021: https://cleanroomtechnology.com/news/article_page/Residue_removal_in_cleanrooms_A_regulatory_overview/161330
- European Medicine Agency (EMA), Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities, November 2014
- European Commission, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use – Annex 15, Qualification and Validation, (2015).
- European Commission, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use – chapter 3, Premises and equipment, (2015).
- European Commission, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use – chapter 5, Production, (2015).
- International Society for Pharmaceutical Engineering (ISPE), Baseline Guide Vol 7: Risk-Based Manufacture of Pharma Products 2nd Edition, July 2017
- Walsh, A., Cleaning Validation for Biologics Can alternative approaches to the Permitted/Acceptable Daily Exposure (PDE/ADE) Be Justified? BioPharm International Supplements, (2015). http://www.biopharminternational.com/cleaning-validation-biologicscan-alternative-approaches-permittedacceptable-daily-exposure-pdeade-be
- Mott, A., Henry, B., Wyman, E., Randall, G., Bellorado, K., Blümel, M., Clark, M.E., Parks, M., Hayes, R., Runkle, S., & Luo W., Methodology for Assessing Product Inactivation During Cleaning Part II: Setting Acceptance Limits of Biopharmaceutical Product Carryover for Equipment Cleaning, Journal of Validation Technology, Vol. 19, Issue 4, (2013).
- European committee for standardization, European standard EN 14885, Chemical disinfectants and antiseptics – Application of European Standards for chemical disinfectants and antiseptics, (2018).
- United States Environmental Protection Agency; Series 810 – Product Performance Test Guidelines, Product Performance Test Guideline, OCSPP 810.2200, Disinfectants for Use on Environmental Surfaces, Guidance for Efficacy Testing (2018)
