


Sommaire
- La campagne on y gagne ! Le travail en mode campagne, une avancée sur la productivité des isolateurs.
- Inspection visuelle : principaux constats des inspections de l’ANSM
- Validation des nettoyages des équipements de production : Contrôle visuel, habilitation du personnel au « visuellement propre »
- A Refresher on Disinfectant Wet Contact Time
- RDM et RDIV de l’UE : Les produits combinés désormais soumis au même degré de surveillance que les dispositifs médicaux autonomes
- Fiabilité de la lignée monocytaire Mono-Mac-6 pour la détection des pyrogènes endotoxiniques et non-endotoxiniques
- Indicateurs biologiques, pousses aléatoires - votre cycle de décontamination est-il réellement en cause ?
- Determining a Strategy for Container Closure Integrity Testing of Sterile Injectable Products
- Efficient Control Strategy enabled by structured Knowledge
- Une solution performante pour les dénombrements en temps réel des colonies sur membranes dans l’analyse microbiologique avec ScanStation
Determining a Strategy for Container Closure Integrity Testing of Sterile Injectable Products
Container closure integrity (CCI) plays an important role in maintaining the sterility and stability of sterile injectable products. The defects which cause a sterile vial to leak are not necessarily defects that will be detected by a visual inspection process. Examples of such defects are defects that are hidden by the crimp, microscopic cracks & scratches in the glass, or temporary defects such as stopper pop-up that result in temporary container leakage.

New regulatory guidance has recently triggered changes in industry best practices in the area of CCI testing (CCIT). This article summarizes the current state of container closure integrity testing in the pharmaceutical and biopharmaceutical industries and outlines possible approaches for developing a CCIT strategy. Concrete industry case studies are presented as examples.
1. Regulatory environment for container closure integrity
Historically, good container closure integrity has been linked to the maintenance of sterility. A container that loses, or does not have, good closure integrity is at risk for microbial contamination. However, the context of container closure integrity has become broader over the years. An increasing number of formulations have some sensitivity to oxygen and need to be packaged under an inert atmosphere. Freeze-dried product requires protection against water vapor and is often packaged at a partial vacuum to help with reconstitution and/or seating of the stopper. Live viral vaccines and gene and cell-based therapies require deep cold storage temperatures (-80 ˚C down to cryo) which can introduce risk to the sealing performance of the packaging components [1].
Increasingly, good container closure integrity is necessary not only for the maintenance of sterility but also to maintain critical headspace gas conditions for stability of the formulation. Note that, quite generally, a container that is gas-tight will also be tight against microbial ingress. Therefore, the requirement to maintain headspace gas conditions imposes higher standards on CCI than the requirement to maintain sterility.
In light of the importance of CCI for product sterility and stability, recent regulatory guidance has placed an increasing emphasis on container closure integrity concepts. The current USP <1207> chapter titled ‘Package Integrity Evaluation – Sterile Products’ was implemented in late 2016 and represents the most thorough guidance document to date on container closure integrity concepts for sterile injectable products. The chapter gives an overview of CCI testing technologies and approaches for a CCI control strategy over the product life cycle. Traditional CCIT methods, such as microbial challenge tests or blue dye ingress tests, are described as methods associated with probabilistic outcomes having some uncertainty in the results which, in turn, makes such methods difficult to quantitatively validate for the detection of critical leaks [2]. The chapter also makes clear that container closure integrity testing should be performed throughout the product life cycle. Deterministic CCIT methods based on non-destructive analytical measurements can be used to generate science-based CCI data that, coupled with a risk-based approach, enables informed decisions about a CCIT strategy in commercial manufacturing.
More recently, a second draft revision of the EU Annex 1 requirements for sterile product manufacturing was released in February, 2020 [3]. Container closure integrity testing was an important topic of discussion for the revision and the draft text contains new requirements for CCI testing in manufacturing. Other world regulatory bodies, Russia and South Korea for example, have also been putting increasing emphasis on CCI control for finished sterile products. It is clear from these recent developments that regulators want to see improved industry practices in the area of CCI testing.
2. Container closure integrity test methods
The USP <1207> chapter provides an overview of CCI testing technologies and categorizes them as being deterministic or probabilistic (see Table 1 below). The chapter emphasizes that this overview of CCI testing technologies is not exhaustive but is a summary of technologies that have been implemented for CCI testing in the pharmaceutical industry and that are described by a body of peer-reviewed literature.
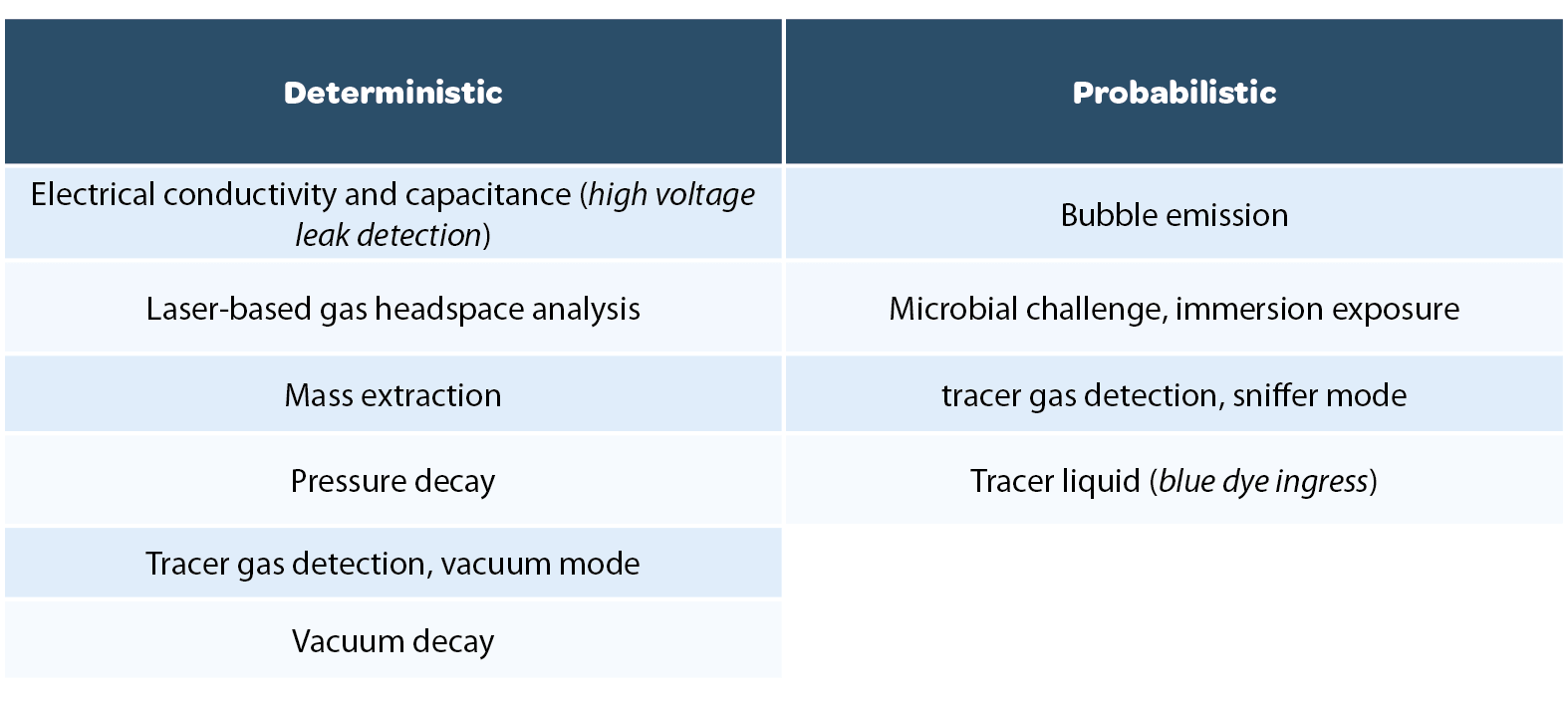
It is important to distinguish between CCI technologies and CCI test methods. Once a leak testing technology has been chosen as the basis for a test method, the chapter emphasizes the need to perform method development studies generating data that demonstrates detection of a critical leak for a specific product container configuration using defined test method parameters: ‘After a methodology has been selected for use, the test equipment operation and performance is qualified. Test method parameters are optimized during method development and confirmed during validation. Thus, a final leak test method is specific to a particular container-closure or product-package system.’ [2] Another point emphasized in the chapter is that ‘no one test is appropriate for all packages or for all leak testing applications’. The chapter and its three sub-sections describe a framework in which appropriate CCI test methodologies are chosen, optimized per product configuration, and a robust validation of the method for detecting a critical leak is performed. In selecting a methodology, ‘deterministic leak test methods are preferred over probabilistic methods when other key method selection criteria permit’. Package integrity data is generated over the product life cycle and serves as input for an ongoing database of CCI data (the package integrity profile) which then serves as a risk management tool to ensure that CCI of finished product meets the product quality requirements. The framework described in the chapter is currently driving changes in industry best practices for CCI testing, including:
• Implementation of a ‘toolbox’ of CCI test methods optimized and chosen on a per product configuration basis rather than the application of a single legacy test method in a one-size-fits-all approach.
• Generation of science-based data in robust CCI product and process studies and in method development & validation studies which demonstrate the detection of a critical leak.
Example: CCIT method development using headspace gas ingress
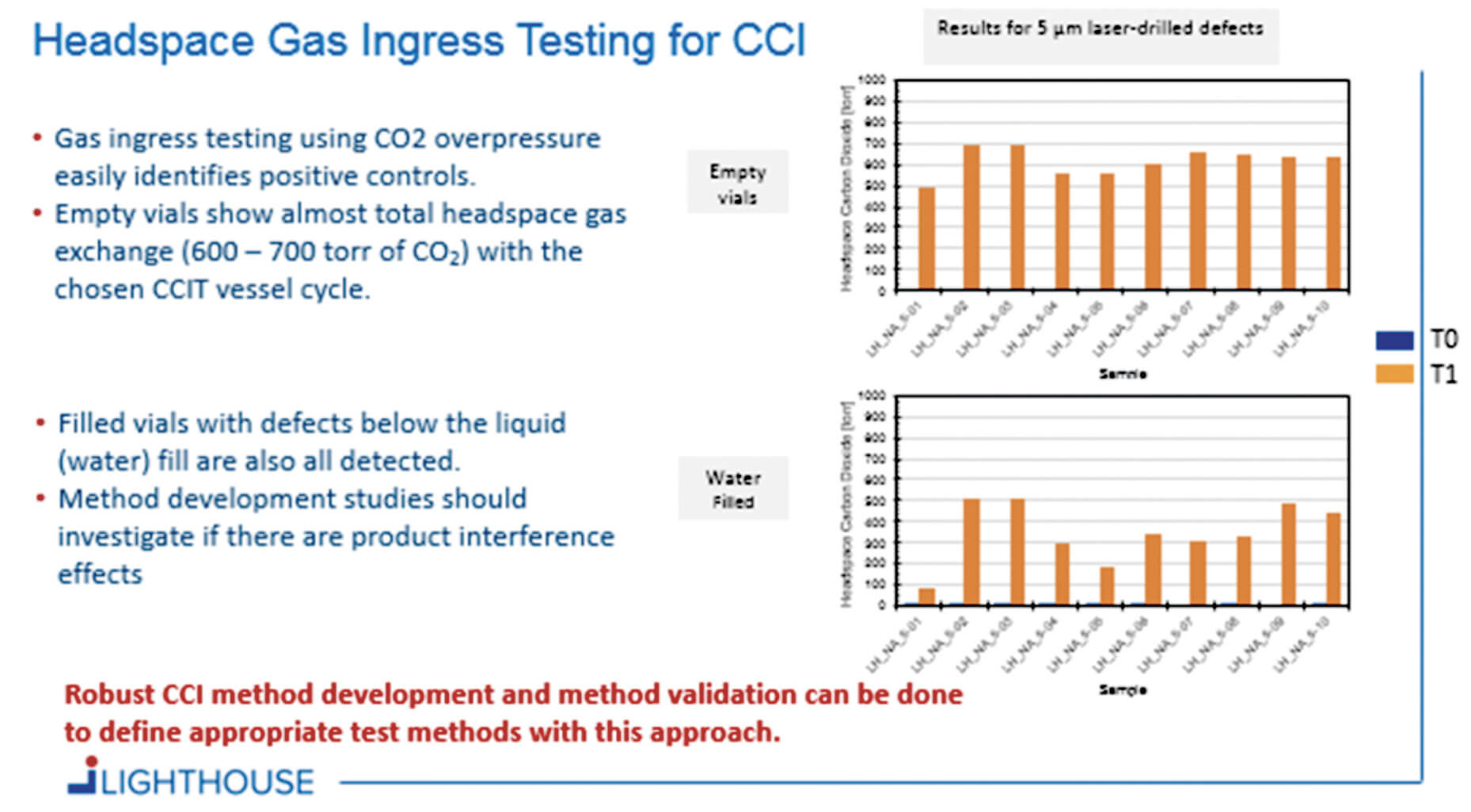
A general approach for CCIT was developed that both resembled and improved upon the blue dye ingress test. A key objective was to develop a method that would reliably detect critical leaks and to move away from the visual, somewhat subjective, inspection by the operator in a blue dye test. For this headspace gas ingress approach, samples are placed into a CCIT vessel which can be pressurized with a tracer gas (e.g., carbon dioxide). If a container has a leak defect, the carbon dioxide gas will ingress into the container (see Figure 1). After the sample conditioning cycle with pressurized CO2, the samples are removed from the vessel and defective vials are identified by using headspace analysis to detect elevated CO2 levels.

Several tests were conducted using filled and empty vials in which 5-μm holes were drilled into the body of the vial and then placed in a CCIT vessel pressurized with CO2. Following a 30-min sample conditioning cycle, a significant amount of CO2 had ingressed into the defective vials.
Figure 2 shows the resulting data of tests conducted with empty vials and vials filled with water. The data illustrates that robust CCI method development and method validation can be done to define appropriate CCIT methods using this approach. Headspace gas ingress testing for CCI uses a ‘gas bath’ instead of a blue-dye molecule bath and detection of any leak is accomplished with a non-destructive analytical headspace measurement.
Statistical sampling and generating science-based CCI data
A topic of current discussion is how much CCI testing should be required, especially for commercial batches of finished sterile products. Despite the general consensus that CCI is a critical quality parameter for finished sterile products, the industry has historically expended much more effort on testing for particle contamination than for CCI. Visual inspection to detect particulate contamination has been a requirement for many years with 100% inspection of finished parenteral products being done manually or by automated inspection platforms. In the context of risk to the patient, a loss of container closure integrity would, in general, be assessed as being just as critical as particle contamination.
The current EU Annex 1 guidelines require 100% leak testing for certain types of product containers: ‘Containers closed by fusion, e.g. glass or plastic ampoules, should be subject to 100% integrity testing’ [4]. This requirement is a result of the fact that the inherent failure rate of the sealing process for these types of containers cannot be sufficiently controlled. The ongoing draft revision of the EU Annex 1 guidelines again states the requirement of 100% integrity testing for fused containers and adds the following requirements for all other types of containers: ‘Samples of containers closed by other methods should be taken and checked for integrity using validated methods. The frequency of testing should be based on the knowledge and experience of the container and closure systems being used. A scientifically valid sampling plan should be utilized. The sample size should be based on information such as supplier approval, packaging component specifications and process knowledge. It should be noted that visual inspection alone is not considered as an acceptable integrity test method.’ [3] There are several interesting discussion points about CCI requirements in this version of the draft revised EU Annex 1 text:
- The frequency of CCI testing in manufacturing is not mandated but should be defined based on ‘knowledge and experience of the container and closure systems’.
- The sample size is also not mandated but should be based on ‘information…and process knowledge’.
If one equates ‘knowledge of the container, closure systems, and process’ to mean ‘data on the container, closure systems, and process’, one comes to the conclusion that scientific CCI studies should be conducted earlier in the product life cycle to justify the CCI testing strategy in manufacturing. In other words, there is an implicit requirement on packaging and process development to generate robust scientific CCI data in support of manufacturing’s efforts to be compliant. If CCI studies generate data showing a high-risk package or process, then a more robust CCI testing strategy should be implemented in manufacturing and vice versa. This follows Quality by Design concepts, namely combining prior knowledge and experimental data from systematic development studies to define a design space for the commercial process ensuring robust quality as well as the implementation of an appropriate control strategy.
Another interesting point of discussion is the language referring to CCI and visual inspection. Some production facilities point to the 100% visual inspection process to justify meeting current CCIT guidance such as the following from the Food and Drug Administration: ‘A container closure system that permits penetration of microorganisms is unsuitable for a sterile product. Any damaged or defective units should be detected, and removed, during inspection of the final sealed product.’ [5] The language of the draft EU Annex 1 revision makes clear that visual inspection is not considered to be an acceptable integrity test method. If statistical or 100% testing is desired, CCI test methods that enable the testing of larger sample sizes will need to be implemented.
To demonstrate statistical confidence in the process requires the generation of statistical CCI data. However, an argument could be made that a better place to do this in the product life cycle is in process development and scale-up rather than in manufacturing. The guidance provided in USP <1207> to collect package integrity data throughout the product life cycle to create a package integrity profile database implies an approach in which a significant amount of CCI data is generated outside of the manufacturing environment. The generation of robust CCI data providing knowledge of the container and closure system and the effects of the process on CCI, which then gives guidance to a CCIT strategy in manufacturing, is exactly what is implied in the draft revised EU Annex 1 text as previously discussed. The schematic below outlines a possible approach to generating CCI data that enables the design of an appropriate CCI testing program in manufacturing.

After validation of the fundamental closure system, data needs to be generated to understand if the process introduces risk to CCI. To gain statistical confidence in the process, it would be necessary to perform testing on statistical sample sets. This in turn will require the use of non-destructive deterministic test methods because the probabilistic legacy test methods (blue dye and microbial ingress testing) have limited throughput capability. Testing could be done on either a pilot scale or with test and engineering batches from the manufacturing environment. Once a baseline failure rate has been established, process controls could be implemented to improve the process, if necessary. Product from the improved process would be tested to quantify the residual risk to CCI after which a decision could be made for an appropriate testing strategy in manufacturing. Packages and processes having a high inherent failure rate that is difficult to control would require a heavier inspection process and vice versa. In this way, the decision for an inspection process design is driven by science-based statistically relevant data.
Example: Data-driven CCI testing strategy in manufacturing
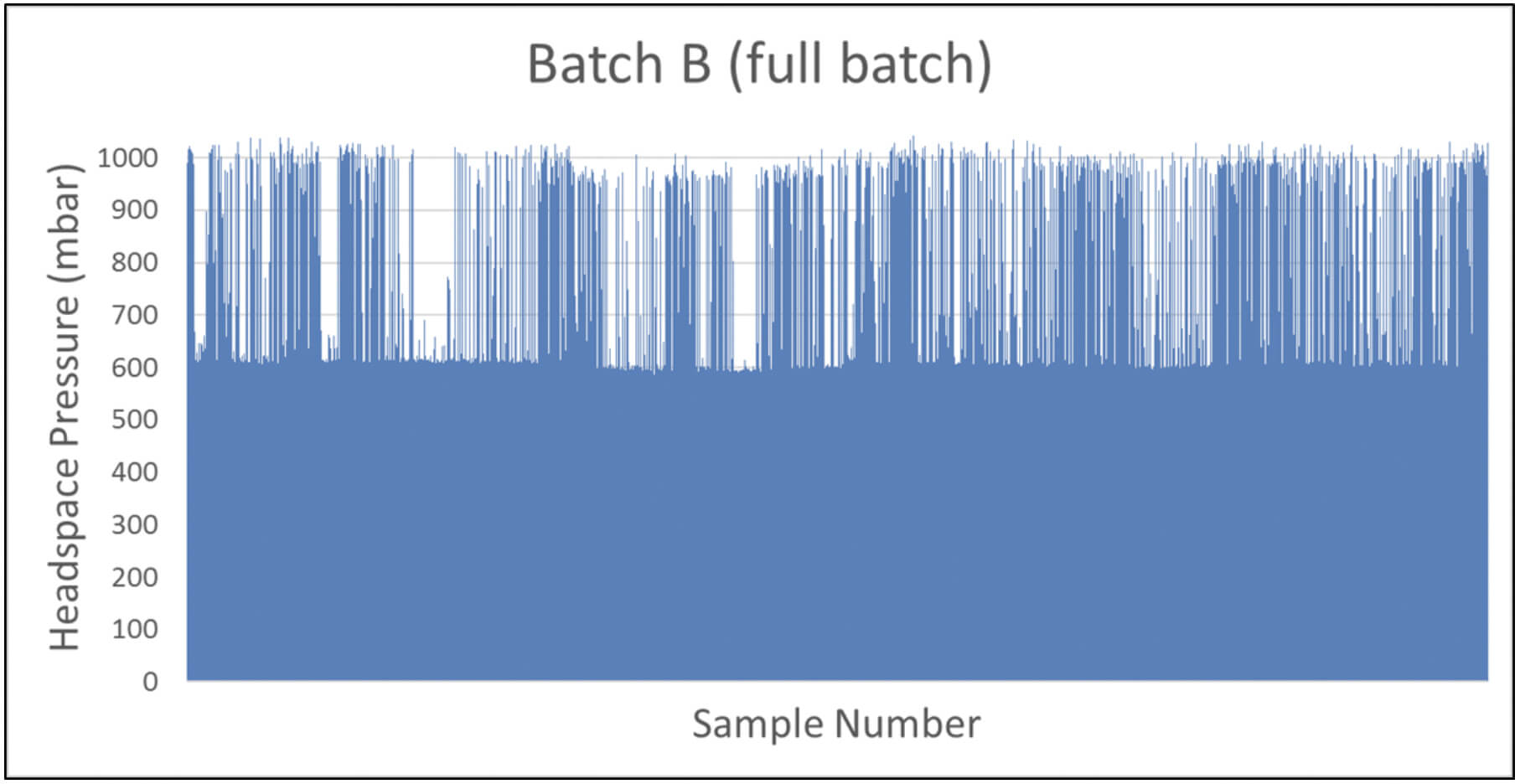
A manufacturer of cytotoxic freeze-dried products identified a batch with potential CCI issues. The batch of 11,000 product vials was put into quarantine. A CCI testing strategy of random sampling based on headspace analysis was implemented. Defective vials were identified by elevated headspace oxygen levels and loss of vacuum as measured by headspace analysis (the product specification was to seal the freeze-dried vials at 600 mbar of nitrogen). Figure 3 shows the headspace oxygen and headspace pressure results of a few hundred samples.
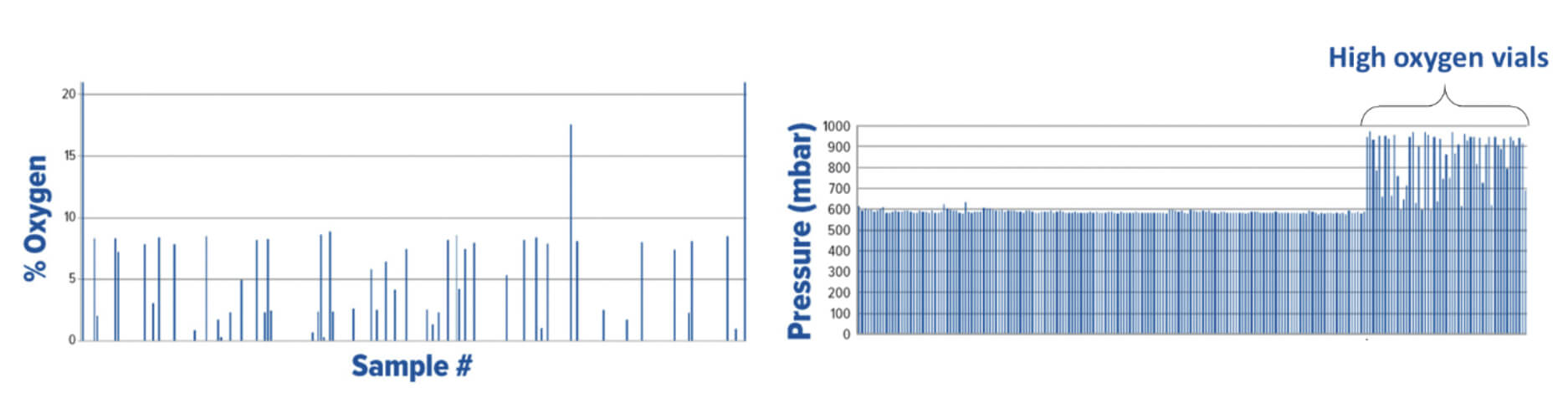
Because of the large number of CCI failures detected with random sampling, an AQL inspection was implemented for the problem batch (Batch B) as well as for the batches produced before and after (Batch A and Batch C, respectively). These results are shown in Figure 4.
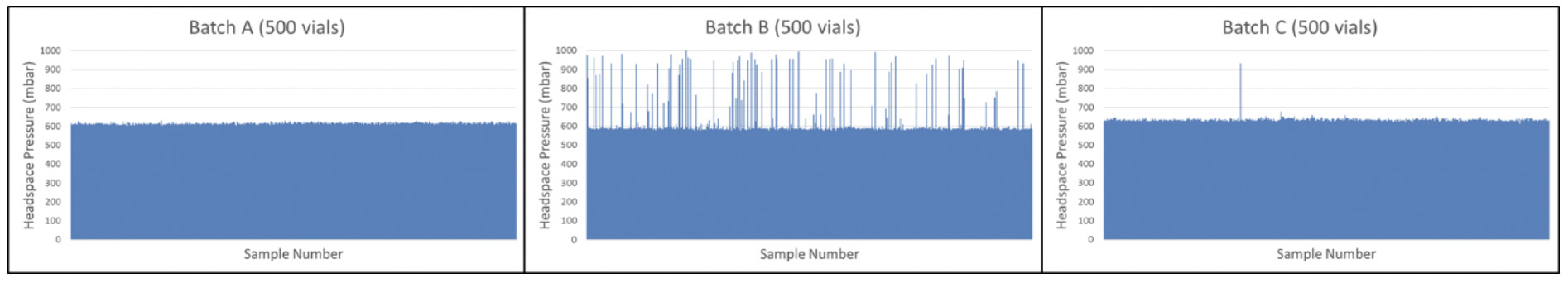
The results of the statistical AQL inspection gave extra insight showing that the problem batch (Batch B) seemed to be an isolated problem batch – 15% of the product vials in Batch B suffered from CCI issues compared with 0% and 0.4% of Batch A and Batch C, respectively. A root cause investigation was started and it was decided to perform 100% CCI inspection of Batch B based on headspace pressure analysis to reject all defective vials from the batch. Results of the 100% CCI inspection are shown in Figure 5 and confirm the conclusion of the AQL inspection that a significant portion of the batch suffered from CCI issues. The data also revealed some product vials suffering from full leaks (full gas exchange and full loss of vacuum) and some product vials suffering from partial leaks (partial gas exchange and partial loss of vacuum). This identified the root cause to be a problem with the stoppering process in the lyo chamber – some vials were leaking coming out of the lyo chamber and were sealed by the capping and crimping process before suffering a full loss of underpressure.
Summary
The current environment for CCI testing of sterile injectable products is evolving. New regulatory guidance recognizes CCI as a quality parameter that is critical for the maintenance of both the sterility and the stability of finished sterile products. New concepts introduced in the regulatory guidance are changing industry best practices and include the following:
- Generate science-based CCI data throughout the product life cycle to build up a package integrity profile database that can be used as input for risk management.
- When possible, use deterministic CCI test methods that have been validated to detect a critical leak.
- There is no one-size-fits-all CCI test; a toolbox of CCI testing technologies that can be optimized on a per product package configuration is necessary for a robust CCIT program.
Because industry best practices will be evolving as the impact of the new guidance becomes clearer, a certain amount of uncertainty in CCIT best practices is to be expected in the near term. However, a general approach that includes a) the implementation of validated deterministic CCIT methods and b) the increased generation of science-based CCI data to enable informed risk assessments, will help prepare the industry for the future.
Partager l’article


Derek Duncan – LightHouse Instruments
Dr. Duncan began his career at the Dutch Institute for Atomic & Molecular Physics in Amsterdam. He moved into industry holding Product & Application Development positions. Currently at LIGHTHOUSE since 2003, Dr. Duncan is responsible for developing applications for process monitoring and finished product inspection. These include using headspace analysis for 100% container closure integrity testing, lyo chamber moisture mapping, and automated media fill inspection.
dduncan@lighthouseinstruments.com
References
[1] Zuleger, B.; Werner, U.; Kort, A.; Glowienka, R.; Wehnes, E.; Duncan, D. Container/Closure Integrity Testing and the Identification of a Suitable Vial/Stopper Combination for Low-Temperature Storage at –80 °C. PDA J. Pharm. Sci. Technol. 2012, 66 (1), 453–465.
[2] U.S. Pharmacopoeia. USP 40 <1207>. Sterile Product Packaging – Integrity Evaluation. United States Pharmacopoeial Convention, Inc.: Rockville, MD, 2017.
[3] EU Annex 1 Revision, February 20th, 2020:
https://ec.europa.eu/health/sites/health/files/files/gmp/2020_annex1ps_sterile_medicinal_products_en.pdf.
[4] European Commission. EudraLex. The Rules Governing Medicinal Products in the European Union. Volume 4: EU Guidelines to Good Manufacturing Practice-Medicinal Products for Human and Veterinary Use. Annex 1Manufacture of Sterile Medicinal Products. Brussels, Belgium: European Commission; 2009.
[5] FDA Guidance for Industry: Sterile Drug Products Produced By Aseptic Processing – Current Good Manufacturing Practice, September 2004 (FDA, Rockville, MD)
